
Upregulation of kazrin F by miR-186 suppresses apoptosis but promotes epithelial-mesenchymal transition to contribute to malignancy in human cervical cancer cells
Introduction
Cervical cancer (CC) ranks as the fourth leading cause of cancer-related death among women worldwide, and the molecular pathogenesis of CC is not well understood (1,2). Illuminating the regulatory molecular network involved in pathogenesis will help to identify biomarkers for the development of clinical diagnosis and treatment approaches.
Kazrin is a widely expressed, evolutionarily conserved cytoplasmic protein. Loss of kazrin in Xenopus tropicalis embryos causes defects in cell adhesion that affect axial elongation, notochord differentiation and epidermal morphogenesis (3). Kazrin, acting as a novel component of desmosomes, is a periplakin interactor (4), and can induce changes in cell shape and stimulate terminal differentiation (5). The kazrin protein has various isoforms, of which kazrin A, B, C and D are associated with cell junctions and mouse and Xenopus embryonic development, while kazrin E is proved to interact with microtubules and involved in epidermal differentiation and desmosome formation. Our previous work identified another new isoform, kazrin F, that has anti-apoptotic effects on a human glioma cell line (6). However, whether kazrin F is involved in tumorigenesis is not clear.
MicroRNAs (miRNAs) are a large group of small non-coding RNAs of ~22 nucleotides in length that bind to the 3'-untranslated region (3'UTR) of most protein-coding transcripts to induce translational repression, mRNA deadenylation and mRNA decay (7,8). miRNAs have fundamental effects on the regulation of intracellular processes, and their importance during malignant transformation and metastasis is becoming increasingly well understood. Many studies have revealed that altered expression of miRNAs contributes to the initiation and development of cancer (9-11). Recently, reports have demonstrated that miR-186 acts as a tumor suppressor and its expression is downregulated in various human malignancies, including bladder cancer (12), non-small cell lung carcinoma (13), prostate cancer (14), endometrial cancer (15), and medulloblastomas (16). However, the effect of miR-186 on CC remains unknown.
In the current study, we found that kazrin F is upregulated and promotes malignancy. Furthermore, miR-186 targets the 3'UTR of kazrin F and augments its expression to promote oncogenic activity in CC cells.
Materials and methods
Clinical tissue samples and cell culture
Human CC samples were collected with informed consent and confirmed by pathologists of the Sun Yat-Sen University Cancer Center and the study was approved by the Ethic Committee of the Sun Yat-Sen University Cancer Center. The CC cell lines HeLa and C33A were cultured at 37 °C and 5% CO2 in RPMI 1640 or MEM-a medium (GIBCO BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) and 1% PS (100 units/mL penicillin, 100 μg/mL streptomycin). Cells were transfected with plasmid DNA using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol.
Plasmid construction
Our previous work had described the method of the pcDNA3/kazrin F and pSilencer/shR-kazrin F plasmid construction (6). To construct a pcDNA3/pri-miR-186 vector, the DNA fragment carrying pri-miR-186 was inserted into the pcDNA3 vector using Xho I and BamH I sites, as well as miR-205 and miR-377. We also commercially synthesized an inhibitor of miR-186 (ASO-miR-186) (GenePharm, Shanghai, China) which contained a 2'-O-methyl-modified antisense oligonucleotide directly against miR-186. The sequence is shown in Table 1. The kazrin F 3'UTR containing the target sites of miR-186 and its mutant form were cloned into the downstream of enhanced green fluorescent protein (EGFP) reporter vector pcDNA3/EGFP between the BamH I and EcoR I sites.

Full table
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Twenty-four hours after transfection, 8,000 cells/well for HeLa cells or 5,000 cells/well for C33A cells were plated into 96-well plates, and the MTT assay was performed to detect cell viability. For details, consult Ref (17).
Colony formation assay
Twenty-four hours after transfection, 500 HeLa cells or 350 C33A cells were seeded in 12-well plates per well. The number of colonies with more than 50 cells was counted 13 or 15 d later, after crystal-violet staining.
Apoptosis assay
Eighteen hours after transfection, cells were incubated with 6 μg/mL paclitaxelat 37 °C for an additional 6 h. Then, the apoptosis detection was performed using Annexin V-FITC kit (Vazyme Biotech Co., Ltd., Nanjing, China) following the manufacturer's protocol.
Migration and invasion assays
Cells were transfected with pFlag-kazrin F, pshR-kazrin F, pri-miR-186, ASO-miR-186 or their corresponding controls. A polycarbonate membrane with a pore size of 8 μm was coated with or without Matrigel (Clontech, Mountain View, CA, USA) and placed in a 24-well Boyden chamber to analyze the invasion or migration of the tumor cells. The details of the methods are provided in Ref (18).
RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA was extracted from cells with Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The expression levels of miR-186 and kazrin F were analyzed with RT-qPCR using SYBR Premix Ex TaqTM (Promega, Madison, WI, USA). For details, refer to previously described methods (19).
EGFP reporter assay
Cells were seeded in 48-well plates one day before transfection and then co-transfected with pri-miR-186 or ASO-miR-186 and pkF-3'UTR1-EGFP, pkF-3'UTR2-EGFP, and pkF-3'UTR3-EGFP or pkF-3'UTR1(mut)-EGFP, with a separate RFP expression vector, pDsRed2-N1 (Clontech, Mountain View, CA, USA) for normalization. Forty-eight hours after transfection, the EGFP and RFP intensities were measured with a fluorescence spectrophotometer (F4500, Hitachi, Tokyo, Japan).
Statistical analysis
The data were expressed as x±s and analyzed with Student's t-test. Each data set represents three separate experiments. All the statistical analyses were carried out using GraphPad Prism 6.01 (GraphPad Software, Inc., CA, USA). P<0.05 was considered statistically significant.
Results
Dysregulated expression of kazrin F in CC tissues promotes proliferation of HeLa and C33A cells
To explore the level of kazrin F in CC, we examined the expression of kazrin F in 18 pairs of CC tissues and its adjacent non-tumor tissues using RT-qPCR. The results indicated a statistically significant increase in the expression of kazrin F (Figure 1A). To investigate the function of kazrin F in CC cells, we first constructed and validated the efficiency of a kazrin F overexpression vector, pCD3-Flag/kazrin F (pFlag-kazrin F), and the knockdown plasmid pSilencer/shR-kazrin F (pshR-kazrin F) using RT-qPCR and western blot in HeLa and C33A cells (Figure 1B). Then, an MTT assay was performed to determine whether kazrin F influenced cell viability. Compared with the control, HeLa and C33A cells transfected with pFlag-kazrin F showed a significant increase in cell viability. Conversely, HeLa and C33A cells transfected with pshR-kazrin F exhibited decreased cell viability (Figure 1C). The colony formation assay revealed that ectopic expression of kazrin F significantly increased the colony formation rate of HeLa and C33A cells, while the opposite effect was observed in cells with kazrin F knockdown (Figure 1D). To investigate the mechanisms underlying the regulation of cell viability, an apoptosis assay was performed. As shown in Figure 1E, the apoptosis rate was decreased by overexpression of kazrin F but significantly increased when kazrin F was knocked down in HeLa cells. Furthermore, western blot showed that the protein level of cleaved caspase-3 significantly decreased after transfection with kazrin F compared with the control. In contrast, depletion of kazrin F with shRNA increased the protein level of cleaved caspase-3 compared with the control plasmid (Figure 1F). However, we did not observe a significant effect of kazrin F on the cell cycle in HeLa cells (Supplementary Figure S1). These data indicate that kazrin F suppresses apoptosis to promote the proliferation of HeLa and C33A cells.
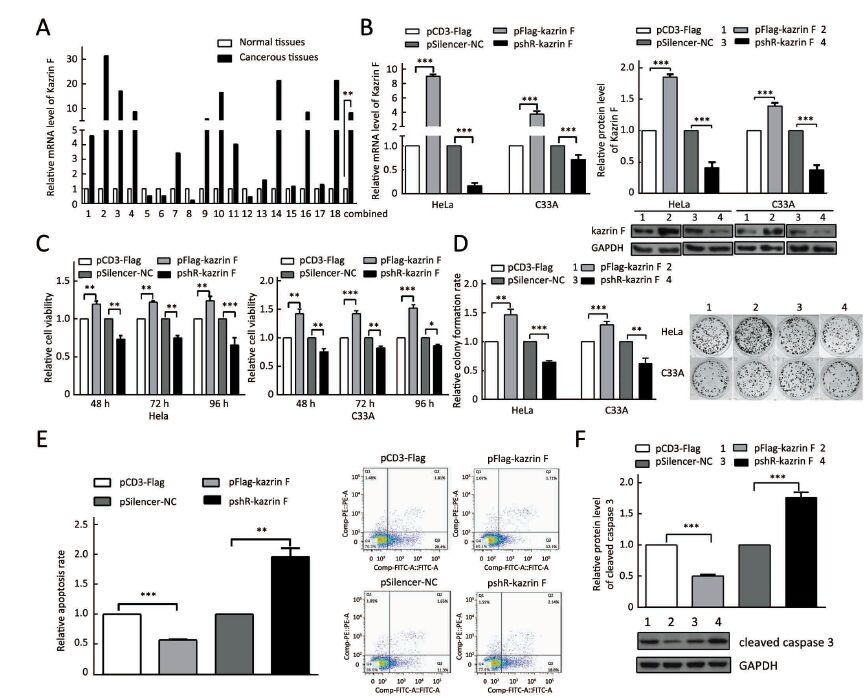
Kazrin F enhances migration and invasion of HeLa and C33A cells
To further explore whether kazrin F affected the migration and invasion ability of HeLa and C33A cells, the transwell assay with or without Matrigel was performed. Overexpression of kazrin F increased the migratory capacity of HeLa and C33A cells, but depletion of kazrin F with shRNA reduced this effect (Figure 2A, left panel). Similar results were observed for the invasion ability of HeLa and C33A cells (Figure 2A, right panel). These data confirm that kazrin F increases the migration and invasion abilities of HeLa and C33A cells.
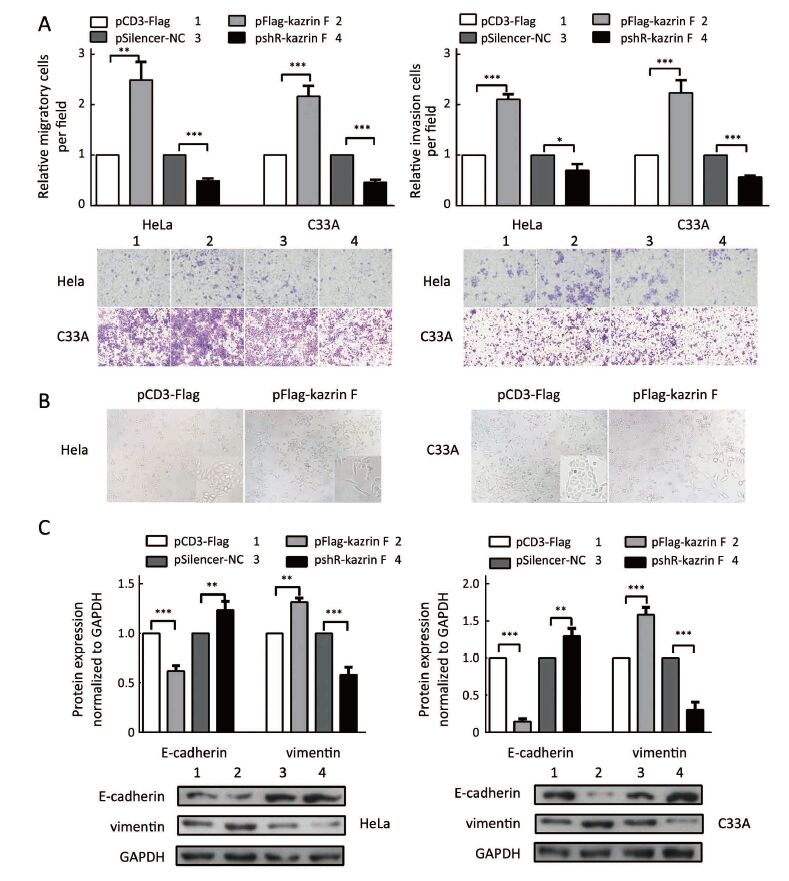
We further ascertained whether kazrin F caused morphological changes in HeLa and C33A cells. As expected, ectopic expression of kazrin F resulted in a shift in cell shape from a cobble-stone-like appearance to a spindle- and fibroblast-like morphology in HeLa and C33A cells, causing substantial morphological changes (Figure 2B). In addition, we examined the expression of molecular markers in epithelial-to-mesenchymal transition (EMT). As shown in Figure 2C, the overexpression of kazrin F increased the E-cadherin protein level but decreased the vimentin protein level in both HeLa and C33A cells. The opposite effects were observed in pshR-kazrin F transfected cells. These results indicate that kazrin F facilitates the epithelial-like/mesenchymal-like phenotype transition as well as migration and invasion in CC cells.
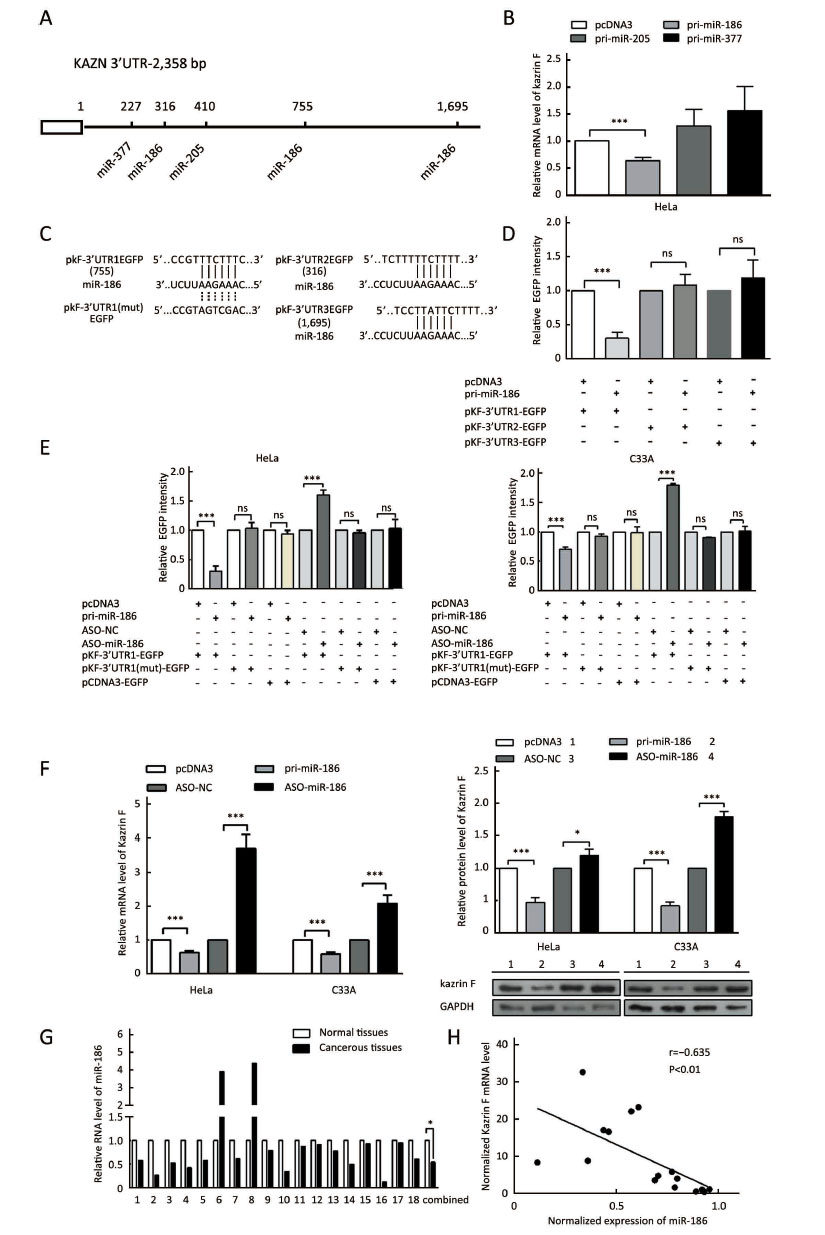
MiR-186 is a direct regulator of kazrin F
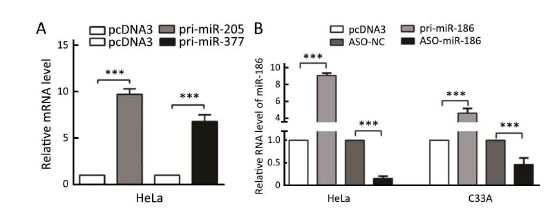
To explore the mechanism of dysregulation of kazrin F in CC, we investigated post-transcriptional regulation by miRNA. We employed the TargetScan, PicTar and miRanda algorithms and predicted three miRNAs that may target the 3'UTR of kazrin F (Figure 3A). To evaluate whether the candidate miRNAs could interfere with kazrin F expression, we increased these miRNA levels in HeLa cells (Supplementary Figure S2). RT-qPCR analyses showed that miR-186 downregulated kazrin F at the mRNA level while miR-205 and miR-377 had no significant effect on kazrin F expression (Figure 3B). Bioinformatics analysis showed that there are three binding sites for miR-186 in the 3'UTR of kazrin F (Figure 3A, C). To further investigate the target sites, we used an EGFP reporter system that utilized the 3'UTR of kazrin F containing the binding sites of miR-186 (named pkF-3'UTR1-EGFP, pkF-3'UTR2-EGFP and pkF-3'UTR3-EGFP) embedded in the downstream region of EGFP in pcDNA3/EGFP. As shown in Figure 3D, only the kF-3'UTR1 EGFP level was decreased by miR-186. Then, we generated some mutant sites in pkF-3'UTR1-EGFP (Figure 3C). Figure 3E showed that EGFP intensity was enhanced after transfection with ASO-miR-186 and suppressed after transfection with pri-miR-186. In contrast, the mutant miR-186 binding sites in the kazrin F 3'UTR abolished the effect of miR-186 on EGFP intensity. In addition, we examined the influence of miR-186 on endogenous kazrin F expression. Consistent with our prediction, ectopic expression of miR-186 decreased the expression of kazrin F at both the mRNA and protein levels in HeLa and C33A cells, whereas blocking miR-186 showed the opposite effects (Figure 3F). Furthermore, RT-qPCR analysis showed that the expression of miR-186 in the 18 pairs of CC tissues was lower in comparison with the corresponding normal tissue (Figure 3G) and showed an inverse correlation with kazrin F expression (Figure 3H). Thus, our results suggest that kazrin F is downregulated by miR-186 at the post-transcriptional level in CC cells.
MiR-186 suppresses proliferation, migration and invasion and regulates EMT-associated molecules in CC cells
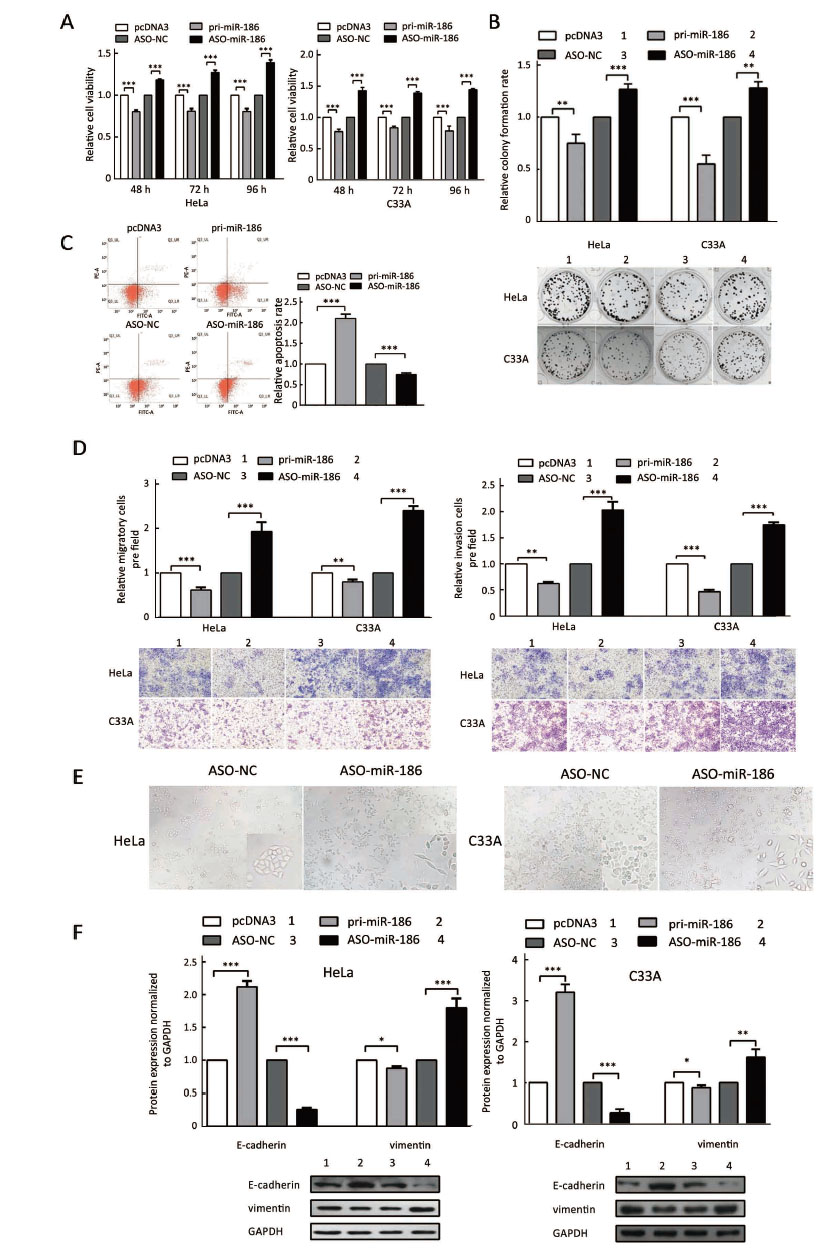
To determine the role of miR-186 in CC cells, HeLa and C33A cells were transfected with pri-miR-186 or ASO-miR-186, and different functional experiments were performed. MTT assay showed that pri-miR-186 significantly decreased cell viability, while ASO-miR-186 increased cell viability in HeLa and C33A cells (Figure 4A). The colony formation assay revealed that overexpression of miR-186 decreased, whereas ASO-miR-186 increased the colony formation rate of HeLa and C33A cells (Figure 4B). The apoptosis assay showed that overexpression of miR-186 increased the apoptosis rate in HeLa cells, while blocking miR-186 decreased the apoptosis rate (Figure 4C). In addition, pri-miR-186 decreased, but ASO-miR-186 promoted, both migration and invasion in HeLa and C33A cells (Figure 4D). Blocking miR-186 in HeLa and C33A cells led to substantial morphological changes (Figure 4E). Furthermore, western blot was used to examine specific EMT-associated molecules. The results showed that E-cadherin levels increased while vimentin levels decreased in miR-186-overexpressing HeLa and C33A cells, while knockdown of miR-186 caused a decrease in E-cadherin but an increase in vimentin (Figure 4F). Taken together, miR-186 may function as a tumor suppressor to suppress cell proliferation, migration and invasion in CC cells.
Ectopic expression of kazrin F restores effects induced by miR-186 in CC cells
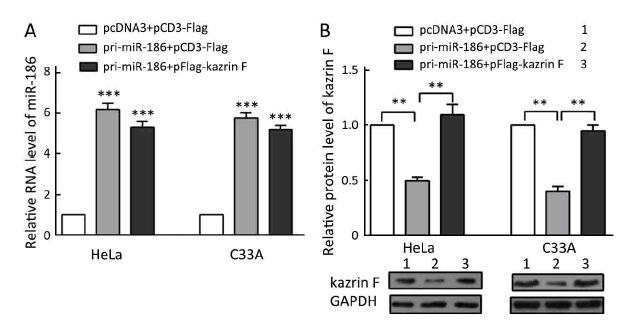
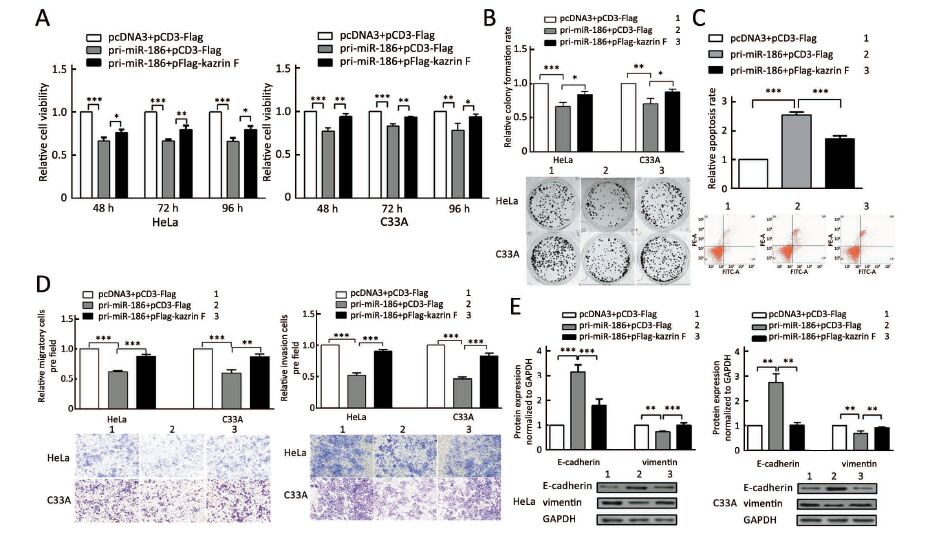
Our data above indicated that miR-186 and kazrin F had opposite effects on the aggressive phenotype of CC cells and that miR-186 downregulated kazrin F expression. We questioned whether the miR-186 phenotype was mediated by kazrin F. We carried out a series of functional rescue experiments. First, we co-transfected pri-miR-186 along with kazrin F into HeLa and C33A cells and determined the expression levels of miR-186 and kazrin F (Supplementary Figure S3). Subsequently, we re-evaluated the effects induced by miR-186 with the presence of kazrin F. As predicted, the ectopic expression of kazrin F counteracted the effects of miR-186 on cell viability, colony formation capacity and apoptosis (Figure 5A, B, C). Moreover, when co-transfected with kazrin F, the effects of miR-186 overexpression on the migration and invasion abilities were significantly impaired (Figure 5D). Furthermore, the expressions of EMT markers in miR-186-expressing cells were restored after overexpression of kazrin F (Figure 5E). These results indicate that the ectopic expression of kazrin F counteracts the inhibition of aggressive phenotypes induced by miR-186 in CC cells.
Discussion
Kazrin has many variants with different functions in cells. Previous studies have shown that kazrin is a constituent of desmosomes and influences intercellular adhesion, growth, development and morphology (20-22). Kazrin acts as a dual regulator of intercellular adhesion and differentiation in keratinocytes and regulates these processes through Rho-dependent and Rho-independent mechanisms (23). Knockdown of kazrin impairs axial elongation, cell differentiation and epidermal morphogenesis in developing Xenopus tropicalis embryos. We previously identified a new isoform named kazrin F and found it plays an important role in regulating cellular apoptosis by interacting with ARC and Bax (6). In this study, we first demonstrated that kazrin F is upregulated in human CC tissues and enhances the proliferation, colony formation, migration and invasion of CC cells. Thus, kazrin F may function as an oncogene in CC.
To explore the mechanism of dysregulation of kazrin F in CC cells, we focused on post-transcriptional regulation by miRNA. We used bioinformatics analysis and predicted three miRNA candidates that may directly target kazrin F. Among the candidates, we chose miR-186 for further study because the other two miRNAs, miR-205 and miR-377, did not significantly influence kazrin F expression. Here, we mainly studied the effect of miR-186 on the expression of kazrin F. miR-186 has been found to influence malignancy in prostate cancer, endometrial cancer and medulloblastomas (14-16). The 3'UTR of kazrin F contained three potential binding sites of miR-186. EGFP reporter assays demonstrated that miR-186 targeted only one binding site to downregulate kazrin F expression. Gain- and loss-of-function assays confirmed that miR-186 downregulates kazrin F expression at the mRNA and protein levels. In addition, RT-qPCR analysis showed that miR-186 expression was inversely correlated with kazrin F expression in clinical CC tissues. Thus, miR-186 targets kazrin F by binding to its 3' UTR and negatively regulates its expression. As we know, like DNA methylation (24), ubiquitination (25), acetylation (26), the regulation of protein expression exists in some processes, which give us great illumination for the future research of kazrin F.
Many studies showed that miRNAs might function as either tumor-suppressing or tumorigenic genes (27-29). miR-186 induced cellular senescence by targeting the alpha subunit of protein kinase CKII in human colorectal cancer cells (30), and inhibited migration and invasion by targeting PTTG1 in human non-small cell lung cancer cells (31). However, the role of miR-186 in CC cells is still unclear. In this study, we showed that miR-186 promoted apoptosis and repressed EMT to facilitate the proliferation, colony formation, migration and invasion of CC cells, suggesting that miR-186 functions as a tumor suppressor in cervical tumorigenesis. The rescue assay showed that the ectopic expression of kazrin F could counteract the inhibition of the aggressive phenotypes induced by miR-186 in CC cells. Together, our data demonstrate that miR-186 functions as a tumor suppressor by downregulating kazrin F in CC cells. In addition, according to database, miR-186 downregulation predicts poor prognosis in patients with pancreatic ductal adenocarcinoma, de novo acute myeloid leukemia, lung adenocarcinoma (32-34), etc. In our study, acting as a tumor suppressor in CC, miR-186 downregulation may correlates with prognosis too. As a new isoform of KAZN, kazrin F has little report. In our study, kazrin F is involved in tumorigenesis and may correlates with prognosis of CC.
Conclusions
The downregulation of miR-186 augments the upregulation of kazrin F, which promotes the proliferation, migration, and invasion processes by repressing apoptosis and promoting EMT in CC cells. Therefore, our results indicate that kazrin F may function as an oncogene and a potential biomarker in CC.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (No. 81572790; 91629302; 31270818) and the Natural Science Foundation of Tianjin (No. 12JCZDJC25100, 14JCYBJC26400, 16JCYBJC42400).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin 2010;60:277–300. [PubMed] DOI:10.3322/caac.20073
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [PubMed] DOI:10.3322/caac.21262
- Sevilla LM, Rana AA, Watt FM, et al. KazrinA is required for axial elongation and epidermal integrity in Xenopus tropicalis. Dev Dyn 2008;237:1718–25. [PubMed] DOI:10.1002/dvdy.21557
- Groot KR, Sevilla LM, Nishi K, et al. Kazrin, a novel periplakin-interacting protein associated with desmosomes and the keratinocyte plasma membrane. J Cell Biol 2004;166:653–9. [PubMed] DOI:10.1083/jcb.200312123
- Nachat R, Cipolat S, Sevilla LM, et al. KazrinE is a desmosome-associated liprin that colocalises with acetylated microtubules. J Cell Sci 2009;122:4035–41. [PubMed] DOI:10.1242/jcs.047266
- Wang Q, Liu M, Li X, et al. Kazrin F is involved in apoptosis and interacts with BAX and ARC. Acta Biochim Biophys Sin (Shanghai) 2009;41:763–72. [PubMed] DOI:10.1093/abbs/gmp065
- Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014;15:509–24. [PubMed] DOI:10.1038/nrm3838
- Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell 2005;122:6–7. [PubMed] DOI:10.1016/j.cell.2005.06.036
- Gao R, Cai C, Gan J, et al. miR-1236 down-regulates alpha-fetoprotein, thus causing PTEN accumulation, which inhibits the PI3K/Akt pathway and malignant phenotype in hepatoma cells. Oncotarget 2015;6:6014–28. [PubMed] DOI:10.18632/oncotarget.3338
- Cao Y, Chen J, Wang D, et al. Upregulated in hepatitis B virus-associated hepatocellular carcinoma cells, miR-331-3p promotes proliferation of hepatocellular carcinoma cells by targeting ING5. Oncotarget 2015;6:38093–106. [PubMed] DOI:10.18632/oncotarget.5642
- Zhang Y, Dai J, Deng H, et al. miR-1228 promotes the proliferation and metastasis of hepatoma cells through a p53 forward feedback loop. Br J Cancer 2015;112:365–74. [PubMed] DOI:10.1038/bjc.2014.593
- Yao K, He L, Gan Y, et al. MiR-186 suppresses the growth and metastasis of bladder cancer by targeting NSBP1. Diagn Pathol 2015;10:146. [PubMed] DOI:10.1186/s13000-015-0372-3
- Cui G, Cui M, Li Y, et al. MiR-186 targets ROCK1 to suppress the growth and metastasis of NSCLC cells. Tumour Biol 2014;35:8933–7. [PubMed] DOI:10.1007/s13277-014-2168-6
- Erdmann K, Kaulke K, Thomae C, et al. Elevated expression of prostate cancer-associated genes is linked to down-regulation of microRNAs. BMC Cancer 2014;14:82. [PubMed] DOI:10.1186/1471-2407-14-82
- Myatt SS, Wang J, Monteiro LJ, et al. Definition of microRNAs that repress expression of the tumor suppressor gene FOXO1 in endometrial cancer. Cancer Res 2010;70:367–77. [PubMed] DOI:10.1158/0008-5472.CAN-09-1891
- Lv SQ, Kim YH, Giulio F, et al. Genetic alterations in microRNAs in medulloblastomas. Brain Pathol 2012;22:230–9. [PubMed] DOI:10.1111/j.1750-3639.2011.00523.x
- Liu RY, Diao CF, Zhang Y, et al. miR-371-5p down-regulates pre mRNA processing factor 4 homolog B (PRPF4B) and facilitates the G1/S transition in human hepatocellular carcinoma cells. Cancer Lett 2013;335:351–60. [PubMed] DOI:10.1016/j.canlet.2013.02.045
- Peng RQ, Wan HY, Li HF, et al. MicroRNA-214 suppresses growth and invasiveness of cervical cancer cells by targeting UDP-N-acetyl-α-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase 7. J Biol Chem 2012;287:14301–9. [PubMed] DOI:10.1074/jbc.M111.337642
- Zhao JL, Zhang L, Guo X, et al. miR-212/132 downregulates SMAD2 expression to suppress the G1/S phase transition of the cell cycle and the epithelial to mesenchymal transition in cervical cancer cells. IUBMB Life 2015;67:380–94. [PubMed] DOI:10.1002/iub.1381
- Chhatriwala MK, Cipolat S, Sevilla LM, et al. Exons 5-15 of kazrin are dispensable for murine epidermal morphogenesis and homeostasis. J Invest Dermatol 2012;132:1977–87. [PubMed] DOI:10.1038/jid.2012.110
- Cho K, Lee M, Gu D, et al. Kazrin, and its binding partners ARVCF- and delta-catenin, are required for Xenopus laevis craniofacial development. Dev Dyn 2011;240:2601–12. [PubMed] DOI:10.1002/dvdy.22721
- Gallicano GI, Foshay K, Pengetnze Y, et al. Dynamics and unexpected localization of the plakin binding protein, kazrin, in mouse eggs and early embryos. Dev Dyn 2005;234:201–14. [PubMed] DOI:10.1002/(ISSN)1097-0177
- Sevilla LM, Nachat R, Groot KR, et al. Kazrin regulates keratinocyte cytoskeletal networks, intercellular junctions and differentiation. J Cell Sci 2008;121:3561–9. [PubMed] DOI:10.1242/jcs.029538
- Zhang PP, Wang XL, Zhao W, et al. DNA methylation-mediated repression of miR-941 enhances lysine (K)-specific demethylase 6B expression in hepatoma cells. J Biol Chem 2014;289:24724–35. [PubMed] DOI:10.1074/jbc.M114.567818
- Lan X, Atanassov BS, Li W, et al. USP44 is an integral component of N-CoR that contributes to gene repression by deubiquitinating histone H2B. Cell Rep 2016;17:2382–93. [PubMed] DOI:10.1016/j.celrep.2016.10.076
- Zhao X, Morikawa Y, Qi F, et al. A novel kinetic model for polysaccharide dissolution during atmospheric acetic acid pretreatment of sugarcane bagasse. Bioresour Technol 2014;151:128–36. [PubMed] DOI:10.1016/j.biortech.2013.10.036
- Ren ZJ, Nong XY, Lv YR, et al. Mir-509-5p joins the Mdm2/p53 feedback loop and regulates cancer cell growth. Cell Death Dis 2014;5:e1387. [PubMed] DOI:10.1038/cddis.2014.327
- Kang HW, Wang F, Wei Q, et al. miR-20a promotes migration and invasion by regulating TNKS2 in human cervical cancer cells. FEBS Lett 2012;586:897–904. [PubMed] DOI:10.1016/j.febslet.2012.02.020
- Zhou J, Tian Y, Li J, et al. miR-206 is down-regulated in breast cancer and inhibits cell proliferation through the up-regulation of cyclinD2. Biochem Biophys Res Commun 2013;433:207–12. [PubMed] DOI:10.1016/j.bbrc.2013.02.084
- Kim SY, Lee YH, Bae YS. MiR-186, miR-216b, miR-337-3p, and miR-760 cooperatively induce cellular senescence by targeting α subunit of protein kinase CKII in human colorectal cancer cells. Biochem Biophys Res Commun 2012;429:173–9. [PubMed] DOI:10.1016/j.bbrc.2012.10.117
- Li H, Yin C, Zhang B, et al. PTTG1 promotes migration and invasion of human non-small cell lung cancer cells and is modulated by miR-186. Carcinogenesis 2013;34:2145–55. [PubMed] DOI:10.1093/carcin/bgt158
- Zhang ZL, Bai ZH, Wang XB, et al. miR-186 and 326 predict the prognosis of pancreatic ductal adenocarcinoma and affect the proliferation and migration of cancer cells. PLoS One 2015;10:e0118814. [PubMed] DOI:10.1371/journal.pone.0118814
- Zhang TJ, Wang YX, Yang DQ, et al. Down-regulation of miR-186 correlates with poor survival in de novo acute myeloid leukemia. Clin Lab 2016;62:113–20. [PubMed]
- Cai J, Wu J, Zhang H, et al. miR-186 downregulation correlates with poor survival in lung adenocarcinoma, where it interferes with cell-cycle regulation. Cancer Res 2013;73:756–66. [PubMed] DOI:10.1158/0008-5472.CAN-12-2651