

Intrinsic apoptotic pathway and G2/M cell cycle arrest involved in tubeimoside I-induced EC109 cell death
Introduction
Esophageal cancer (EC) is one of the most lethal cancers and ranks as the sixth leading cause of cancer-related death throughout the world (1). EC, arising from the inner layer of esophagus, represents two histological subtypes, including esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC) (2). Generally, most of EC in developing countries is ESCC, while EAC is more often in Western countries, which has the distinguishing etiologies and results in different therapeutics and prognosis. Moreover, ESCC is the major predominant cancer of esophagus, accounting for more than 90% of diagnosed cases, especially in China (3). Although multimodal therapies and the optimal approaches are still controversial issues, chemotherapy or chemoradiotherapy is also applied widely in clinic, particularly in unresectable EC (1).
The tuber of Bolbostemma paniculatum (Maxim.) Franquet (Cucurbitaceae), Chinese name as Tu Bei Mu (TBM), is a traditional folk medicine, and due to the heat and toxin clearing action, it has been applied in the treatment of mastitis, breast abscess, tubercular lymphadenitis and cancers, especially EC. Tubeimoside I (TBMS1) (Figure 1), a triterpenoid saponin, is the main component of TBM with anticancer activity. However, the detailed mechanisms and intracellular targets underlying TBMS1-induced EC cells toxicity remain elusive.
Mitochondrion is a key organelle inducing cell apoptosis in many tumor treatments. Increasing investigations have found that mitochondrial dysfunction is an effective way to suppress tumor progression, and numerous anticancer drugs evoke cancer cell death through mitochondrial dysfunction. The classical mitochondria- related apoptosis is to transport death signals via Bcl-2 family proteins, leading depletion of outer membrane potential, release of proteins residing in mitochondrial intermembrane space (MIS) and activation of caspase cascade (4). The proteins leaking out from the MIS include cytochrome c, endonuclease G and apoptosis inducing factor (AIF) (5).
Deregulation of cell cycle regulators in different levels is detected in many human cancers. At the same time, cell cycle checkpoints are usually the potential anticancer chemotherapeutic targets. There are two conversed types of anticancer agents evoked by G2/M checkpoint deregulation (6). One affects the abrogation of G2/M checkpoint, making the damaged cells to mitosis stage and further inducing apoptosis (7). Another way is to promote the G2/M checkpoint, subsequently inducing apoptosis (8). Increasing evidence showed that many anticancer compounds trigger apoptosis, accompanying G2/M cell cycle arrest.
In the present study, we applied proteomics approach to predict the cytotoxicity mechanism of TBMS1 in ESCC cells (9). Our findings indicated that TBMS1 exhibited its cytotoxicity associated with mitochondrial dysfunction and G2/M cell cycle arrest. Further biochemical study revealed that caspase-3 and -9 were activated and AIF released. In addition, cyclin B/cdc2 complex and P21 were proved to play a critical role in TBMS1-induced EC109 cell G2/M arrest. Our findings may provide a rationale for the further investigation of TBMS1 in ESCC treatment.

Materials and methods
Materials
TBMS1 was purchased from National Institute for the Control of Pharmaceutical and Biological Products (China), dissolved in phosphate buffered saline (PBS) and stored at –20 °C. JC-1 was purchased from Molecular Probes (Invitrogen, CA, USA). 4',6-diamidino-2-phenylindole (DAPI) was brought from Roche (Mannheim, Germany). Subcellular proteome extract kit (S-PEK) was obtained from CalBiochem (San Diego, CA, USA). All other chemicals, except otherwise noted, were purchased from Sigma.
Cell culture and treatment
EC109 cells were cultured in DMEM (High Glucose) supplemented with 10% fetal bovine serum (FBS) under standard culture condition. At 80% confluence, cultures were treated with desired TBMS1 for cell growth and cell cycle analysis.
Cytotoxicity assay
Cytotoxicity was evaluated by MTT assay. Cells were suspended at 1×104 cells/well and 200 µL of suspension was plated onto each well of a 96-well plate. After 24 h, the medium was replaced by various concentrations of TBMS1. At the end of treatment, the medium was removed, and 20 µL 5 mg/mL MTT in DMEM medium was added. The cells were further incubated in 5% CO2 at 37 °C for 4 h. Formazan was solubilized with 100 µL dimethylsulfoxide (DMSO) and measured at 595 nm.
Colony formation efficiency analysis
The assay was performed in 6-well plates with a bottom layer containing 0.6% agar in DMEM containing 20% FBS. Cells were seeded at a concentration of 5,000/mL in top layer with 0.3% agar, 20% FBS, and treated with desired doses of TBMS1. The plates were incubated at 37 °C for 14 d and the medium with or without TBMS1 was changed every three days. The number of colonies was counted with a microscope and showed as  with three independent assays.
with three independent assays.
DNA laddering
DNA laddering was analyzed by Lei’s protocol (9). Equal DNA extractions from treated or untreated EC109 cells were run on 1% agarose gel, and bands were detected by ethidium bromide staining.
Flow cytometry analysis of cell cycle distribution
EC109 cells were treated with desired doses of TBMS1 for 24 h. At the end of each experiment, 1×106 EC109 cells were harvested, stained with propidium iodide (PI) and evaluated by FACStar Plus flow cytometer. Cell cycle was distributed via the WinMDI 2.9 software.
Confocal microscopic analysis
EC109 cells were attached on cover slips and then exposed to TBMS1 for indicated time. Corresponding primary and fluorescein isothiocyanate (FITC)-conjugated secondary antibodies were used to stain the specific proteins. DAPI was used to stain the nucleus (blue). Mito Tracker red (Molecular Probes, Invitrogen) was performed to dye mitochondria. Corresponding primary and FITC-conjugated secondary antibodies were used to stain AIF and P21 proteins (green). Confocal laser-scanning immunofluorescent microscopy analysis was applied on Zeiss LSM510 Confocal Microscope System (Carl Zeiss, Thornwood, NY).
Two-dimensional electrophoresis (2-DE) and mass spectrometry (MS) analysis
2-DE was performed with GE IPGphor IEF and Ettan Dalt Six electrophoresis units by following protocol described previously. Image analysis was carried out with the ImageMaster 2D Elite software (GE Healthcare, UK). Only those protein spots showing reproducible alterations in three independent experiments (over 2.0-fold up- or down-regulation) were selected for tryptic in-gel digestion and MS analysis. Peptide mass spectra were obtained on an ABI 4800-plus MALDI TOF/TOF mass spectrometer in a reflector positive ion mode with average 1,500 laser shots per spectrum. Peptide ion masses were internally calibrated using trypsin autolytic peptides at m/z 842.51 and 2,211.10. TOF/TOF tandem MS fragmentation spectra were acquired in a data-dependent fashion based on the MALDI-TOF peptide mass map for each protein, and 10 most abundant ions present were selected in each sample (excluding trypsin autolytic peptides and other known background ions). All these data were processed by using the GPS Explorer software (V3.6, Applied Biosystems). MASCOT was used in searching for protein identification by NCBInr protein database. The mass tolerance was set at 75 ppm. Species search was limited to human.
Western immunoblotting
Total cell lysates were denatured with sample loading buffer and then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for protein separation. After transferred onto membranes, the proteins were probed with corresponding antibodies and detected by ECL detection reagents (GE, UK). The primary antibodies for cdc2, PARP and P21 were purchased from Cell Signaling (Beverly, MA, USA). Primary antibodies for cyclin B1, Bcl-2 and Bax were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-cdc2 was from Abcam. Antibodies for β-actin and secondary anti-mouse and rabbit antibodies were from Sigma (USA).
Statistical analysis
Statistical analysis was done through Excel data analysis using a two tailed Student’s t-test and P<0.05 was considered statistically significant. Data are expressed as of triplicate samples, and the reproducibility was confirmed in three separate experiments.
Results
EC109 cells show apoptotic characteristics with TBMS1 treatment
After treatment with indicated concentration of TBMS1 (Figure 1) for 24 or 48 h, we concluded that IC50 of TBMS1 to EC109 was almost 45 and 35 µmol/L at 24 and 48 h, respectively (Figure 2A,B). DNA laddering was analyzed after 30 and 45 µmol/L TBMS1 administration for 12 h in EC109 cells. The degradation of cellular DNA elevated in a dose-dependent manner (Figure 2C). In addition, typical apoptotic morphological changes of EC109 cells were detected by DAPI staining (Figure 2D). In Figure 2D, TBMS1-induced chromatin condensation was clearly detectable after TBMS1 treatment and a large amount of apoptotic bodies particularly appeared after 24 h. The percentage of apoptotic cells increased by about 13% and 40% over untreated EC109 cells, after 30 and 45 µmol/L TBMS1 treatment for 24 h, respectively. All above data implicated that TBMS1 induced obvious apoptosis in EC109 cells.
 of triplicate experiments (×320); *, P<0.05 compared with untreated EC109 cells
of triplicate experiments (×320); *, P<0.05 compared with untreated EC109 cellsCellular proteins alter in response to TBMS1 administration
S-PEK kit was used to separate the total proteins from EC109 into mainly 3 different parts, including cytoplasm, membrane and nucleus fractions (Figure 3A). Figure 3A shows the representative 2-DE images for nuclear proteins extracted from EC109 cells treated and untreated with 45 µmol/L TBMS1 for 24 h. More than 1,000 protein spots were detected from MW 6-200 kDa and pI 3-10. The altered spots were exhibited in Figure 3B. The spots with great differences (up- or down-regulation over 2-fold) were selected to run MS. Finally, we identified 10 proteins and their spot number, ID, MW/pI values, fold difference, scores and main function are listed in Table 1. These altered proteins are up-regulation of prohibitin (PHB), Tu translation elongation factor, mitochondrial (TUFM) and annexin A2; and down-regulation of β-tubulin, γ-actin, heterogeneous nuclear ribonucleoprotein K (HNRNPK), nucleophosmin 1 (NPM1), lamin B2 (LMNB2), aldolase A and histone H1.

Full Table
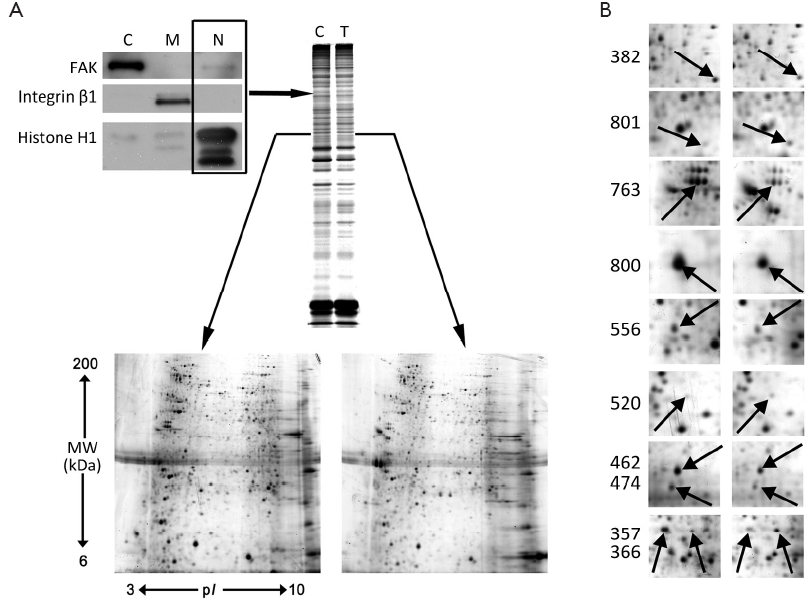
TBMS1 causes mitochondrial dysfunction
Considering that mitochondria were a kind of important organelles initiating apoptosis and proteomic changes related to glycolytic enzyme and mitochondrial stress, we further evaluated the function and corresponding markers of mitochondria-associated proteins. The activity of caspase-3 and caspase-9 was firstly estimated (Figure 4A). Then, mitochondrial transmembrane potential (ΔΨm) was measured to detect the mitochondrial integrity by JC-1 staining (Figure 4B). The activation of caspase-3 and caspase-9 was observed upon TBMS1 treatment with different concentrations and the depletion of ΔΨm exhibited apparently under 45 µmol/L TBMS1 for 6 h. The mitochondrial cell death pathway is regulated by Bcl-2 family. Particularly, the ratio of Bax (pro-apoptotic member) to Bcl-2 (antiapoptotic member) is critical for MIS molecule release and the following caspase activation. Western blot analysis indicated that increased Bax expression was concurrent with suppressed Bcl-2 in a dose-dependent manner (Figure 4C). In addition, fluorescent analysis of AIF revealed that after 45 µmol/L TBMS1 exposure for 12 h, AIF released from mitochondria to cytoplasm, even into nucleus (Figure 4D). These results confirmed that TBMS1 evoked mitochondrial dysfunction-related apoptosis in EC109 cells.

Modulation of G2/M cell cycle regulators by TBMS1
Proteomic alteration also gave us a clue that TBMS1 affects the proliferation of EC109. The soft agar colony formation assay was applied to monitor anchorage-independent growth, measuring proliferation in a semisolid culture media. In the present study, after TBMS1 treatment for 14 d, the number of colonies formed was significantly fewer than the untreated cells (P<0.001) (Figure 5A). Figure 5A clearly shows that TBMS1 caused a concentration-dependent reduction of colony formation, with 54.7% and 21.9% colonies corresponding to the treatment with 30 and 45 µmol/L TBMS1, respectively. Further, we detected the cell cycle distribution via flow cytometry. Figure 5B,C showed that the treatment with 45 µmol/L TBMS1 for 12 and 24 h resulted in an increased population of G2/M phase cells, resulting in 15-24% cell increase in G2/M phase.
Three regulatory molecules, cyclin-dependent kinases (CDKs), cyclins and cyclin-dependent kinase inhibitors (CDKIs), determine the cell cycle progression by controlling the cell cycle. CDK-cyclin complexes are the ultimate target of cell cycle transition. Western blot analysis in Figure 5D showed a strong decrease of cdc2 and cyclin B1 in both cytoplasm and nucleus fraction. Other negative regulators of CDKs are a family of inhibitory proteins known as CDKIs, including Cip1/P21 and Kip1/P27. Our results revealed that TBMS1 treatment caused a strong overexpression of Cip1/P21 in cytoplasm and nucleus (Figure 5D). We also detected the decrease of Cip1/P21 with fluorescent analysis (Figure 5E), verifying that Cip1/P21 accumulated to the nucleus to interrupt the cell cycle progression. In this respect, our data suggest that TBMS1 affected cdc2/cyclin B1 complex pathway and P21 expression and location, contributing to the G2/M arrest.

Discussion
TBM is the key herb constituent in the Chinese folk medicine to treat cancer for thousands of years. Numerous evidences indicated that TBMS1 is the crucial component of TBM with anticancer effect on several cancer cell lines. Especially, our previous results demonstrated that TBMS1 could induce death of HeLa, HepG2, A549 and HL-60 cells (data not shown). In addition, earlier reports demonstrated that TBMS1 has low toxicity in vitro and in vivo (10). This conclusion coincided with that triterpenoid saponins extracted from traditional Chinese herbal medicines have been shown low cytotoxicity to normal cells. Particularly, based on a long term clinical experience in traditional Chinese herb medicine for EC treatment, we suggested that TBMS1 could be used in chemotherapy and has low toxicity. Because ECSS is a main subtype of EC in China, here we choose EC109 as the cell model. In the present investigation, the evaluation of the mechanism underlying TBMS1-induced EC109 cell death could contribute to the development of TBMS1 as a promising anticancer agent in ECSS treatment.
From previous results, we have known that there is a broad spectrum of carcinoma cell species for TBMS1-induced cytotoxicity, including HeLa, A549, HL-60, SGC-7901, PC-3 and Bel-7402 (11). Here, we examined the effect on EC109, showing the dose- and time-dependent cytotoxicity. Then, we evaluated TBMS1-induced alteration of nucleus morphology and DNA fragmentation, which indicated TBMS1-associated apoptosis. To give an insightful understanding of TBMS1’s effects on EC109, nuclear comparative proteomic analysis was further carried out. Compared to the total protein profiling, this method will reveal more detailed information from nucleus, implicating several involved cell signaling pathways. The altered spots proteins are listed in details as follows.
LMNB2 is believed to be critical for eukaryotic cells in DNA replication and in the formation of the mitotic spindle. The absence or down-regulation of LMNB2 in HeLa cells was demonstrated to arrest cell growth and lead to apoptosis (12). β-tubulin is one of the essential components of microtubule and its dynamic alteration has been proposed as a target of anticancer drugs, inducing mitotic arrest of cells in the G2/M stage (13). HNRNPK belongs to the subfamily of ubiquitously expressed heterogeneous nuclear ribonucleoproteins (hnRNPs). Several experiments verified that HNRNPK acted as a cofactor for P53 in response to DNA damage (14-16). Knockdown of HNRNPK by siRNA inhibited pancreatic cancer cell growth and colony formation, implying its function during cell cycle progression (15).
Of note, all these altered proteins in current study are located in nucleus; however, most of them participate in the integration cell signaling process between nucleuses and cytoplasm or mitochondria, such as TUFM, NPM1, PHB, aldolase A and annexin A2. TUFM plays a central role in protein translation in mitochondria. Thus, in our experiment, down-regulation of TUFM will directly influence on the synthesis of mtDNA-encoded proteins, implicating the mitochondrial damage. Moreover, over-expression of TUFM was proved to rescue mitochondrial dysfunction (17) and ShRNA-mediated knockdown of TUFM in leukemic cells exhibited the similar outcome to the interrupted mitochondrial translation and antileukemia activity (18). NPM1 is multifunctional nuclear acidic chaperone, which moves between the nucleus and the cytoplasm involving in several processes, including ribosome biogenesis, chromatin remodeling, and mitosis as well as in DNA repair, replication and transcription (19). Expression of NPM1 is weak in normal hepatocytes and has an obvious positive correlation with the increased tumor grading and poor prognosis (20). In addition, suppression of NPM1 expression was found to enhance adriamycin chemosensitivity in adriamycin-resistant human gastric cancer cell line (21). NPM1 also plays an important role in the regulation of the alternate reading frame (ARF)-P53 tumor-suppressor pathway (22). PHB is an evolutionary conserved protein, involved in the regulation of cell growth, proliferation, differentiation, aging, and apoptosis (23,24). It was initially believed to be a chaperone with tumor suppression function that resides in the mitochondrial inner membrane (25); however, increasing evidence revealed that PHB also located in nuclear matrix and carried out its function through being exported from the nucleus to the mitochondria (26). Accumulation of PHB in nucleus may reflect the unsuccessful translocation to mitochondria and dysfunction of mitochondria. Aldolase A is a glycolytic enzyme, existing both in nucleus and cytoplasm (11), even in mitochondrial fraction (11). Knockdown of aldolase A in Ras-transformed NIH-3T3 cells causes inhibition of the cell proliferation, whereas it could be recovered by exogenous aldolase A. Accordingly, knockdown of aldolase A by RNAi was indicated as a potential therapeutic target for cancer treatment (27). Annexin A2 is a pleiotropic protein and plays its role depending on its different location. It involved in diverse cellular processes such as cell motility, endocytosis, cell proliferation, apoptosis and cytoskeleton reorganization (11). Some evidence shows that annexin A2 could interact with PHB for its function (28).
Therefore, based on the above analysis, these proteins mainly contributed to abnormal mitochondrial function and cell proliferation inhibition. Consequently, mitochondria-related function and TBMS1-induced cell proliferation were further evaluated.
Disruption of mitochondrial transmembrane potential and the elevated caspase-9 and -3 activities (Figure 5A,B) provided a powerful hints that intrinsic apoptotic pathway was involved in TBMS1-related EC109 cytotoxicity. The intrinsic apoptotic pathway is initiated mainly by the dysfunction of mitochondria, which is regulated via the Bcl-2 family proteins. In our current study, the apparently increased ratio of Bax to Bcl-2 and the cleaved-PARP confirmed the mitochondria-initiated intrinsic apoptosis. In addition, after mitochondrial transmembrane damage, cytochrome c, Smac/DIABLO and AIF would release from the MIS. Especially, AIF, a mitochondrial flavoprotein, leaked out from MIS, and then translocated from cytoplasm to nucleus during apoptosis, finally leading to chromatin condensation and large scale DNA fragmentation independent of caspases (4,29-31). Therefore, our results implied that TBMS-1 induced mitochondrial intrinsic apoptosis through caspase-dependent and -independent pathways.
Next, we focused on the TBMS1-mediated proliferation and cell growth. TBMS1 inhibited the proliferation of EC109 and arrested the cells in the G2/M phase. Furthermore, we found the increased population of G2/M cells has a great positive relation with elevated apoptotic population (Figure 5B). The heterodimeric complex of cdc2 and cyclin B1 is the ultimate target of G2/M cell cycle. TBMS1 decreased cdc2 protein levels, with a concomitant decrease in cyclin B1 expression in EC109 cells, both in cytoplasm and nucleus fractions.
Besides cyclin and CDK complex, CDKIs are the other key regulators in the cell cycle progression. P21 is not only the principal mediator of G1-phase arrest in response to DNA damage (32), but also participates in the maintenance of cells in G2/M phase through multiple mechanisms (33). Although P21 has a relatively low affinity to cyclin B1-cdc2 complex, it has been demonstrated to contribute to cdc2 inactivation by inhibiting the cdc2 phosphorylation at Thr161 to enforce the G2/M DNA damage checkpoint (34). In current study, P21 was observed not only elevated expression, but also translocated to nuclear (Figure 5D,E), which further demonstrated its function on cyclin B1 and cdc2 complex.
In this project, we explored the alteration of nuclear proteins after TBMS1 exposure to EC109. From the identified protein, mitochondrial dysfunction and cell cycle arrest were speculated to involve in the process of TBMS1-mediated EC109 cell death. Further functional studies demonstrated that TBMS1-induced apoptosis is through mitochondrial intrinsic apoptosis and P21-cdc2/cyclin B1 signaling pathway. In addition, mitochondria-nucleus communication in TBMS1-induced EC109 cytotoxicity excites our attention, and it may be the main role in this process and deserved further investigation.
Acknowledgements
This work was supported by the Natural Science Foundation of Fujian Province of China (No. 2011J05098), the Fundamental Research Funds for the Central Universities (No. 2011121055), Grants from the National Natural Science Foundation of China (No. 81202956), SRF for ROCS, SEM [2011]1568 and NSFC (No. 81102332).
Disclosure: The authors declare no conflict of interest.
References
- Schweigert M, Dubecz A, Stein HJ. Oesophageal cancer-an overview. Nat Rev Gastroenterol Hepatol 2013;10:230-44. [PubMed]
- Pennathur A, Gibson MK, Jobe BA, et al. Oesophageal carcinoma. Lancet 2013;381:400-12. [PubMed]
- Metzger R, Schneider PM, Warnecke-Eberz U, et al. Molecular biology of esophageal cancer. Onkologie 2004;27:200-6. [PubMed]
- Zhai D, Jin C, Huang Z, et al. Differential regulation of Bax and Bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J Biol Chem 2008;283:9580-6. [PubMed]
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev 2001;15:2922-33. [PubMed]
- DiPaola RS. To arrest or not to G(2)-M Cell-cycle arrest: commentary re: A. K. Tyagi et al., Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G(2)-M arrest, and apoptosis. Clin Cancer Res 2002;8:3311-4. [PubMed]
- Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 2004;3:513-9. [PubMed]
- Weir NM, Selvendiran K, Kutala VK, et al. Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer Biol Ther 2007;6:178-84. [PubMed]
- Lei T, He QY, Cai Z, et al. Proteomic analysis of chromium cytotoxicity in cultured rat lung epithelial cells. Proteomics 2008;8:2420-9. [PubMed]
- Yu L, Ma R, Wang Y, et al. Potent anti-tumor activity and low toxicity of tubeimoside 1 isolated from Bolbostemma paniculatum. Planta Med 1994;60:204-8. [PubMed]
- Yin Y, Chen W, Tang C, et al. NF-κB, JNK and p53 pathways are involved in tubeimoside-1-induced apoptosis in HepG2 cells with oxidative stress and G2/M cell cycle arrest. Food Chem Toxicol 2011;49:3046-54. [PubMed]
- Steen RL, Collas P. Mistargeting of B-type lamins at the end of mitosis: implications on cell survival and regulation of lamins A/C expression. J Cell Biol 2001;153:621-6. [PubMed]
- Eisenlöffel C, Schmöle AC, Pews-Davtyan A, et al. Interference of a novel indolylmaleimide with microtubules induces mitotic arrest and apoptosis in human progenitor and cancer cells. Biochem Pharmacol 2013;85:763-71. [PubMed]
- Pelisch F, Pozzi B, Risso G, et al. DNA damage-induced heterogeneous nuclear ribonucleoprotein K sumoylation regulates p53 transcriptional activation. J Biol Chem 2012;287:30789-99. [PubMed]
- Zhou R, Shanas R, Nelson MA, et al. Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int J Cancer 2010;126:395-404. [PubMed]
- Lee SW, Lee MH, Park JH, et al. SUMOylation of hnRNP-K is required for p53-mediated cell-cycle arrest in response to DNA damage. EMBO J 2012;31:4441-52. [PubMed]
- Feuermann M, Francisci S, Rinaldi T, et al. The yeast counterparts of human ‘MELAS’ mutations cause mitochondrial dysfunction that can be rescued by overexpression of the mitochondrial translation factor EF-Tu. EMBO Rep 2003;4:53-8. [PubMed]
- Skrtić M, Sriskanthadevan S, Jhas B, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011;20:674-88. [PubMed]
- Lindström MS. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem Res Int 2011;2011:195209.
- Liu X, Liu D, Qian D, et al. Nucleophosmin (NPM1/B23) interacts with activating transcription factor 5 (ATF5) protein and promotes proteasome- and caspase-dependent ATF5 degradation in hepatocellular carcinoma cells. J Biol Chem 2012;287:19599-609. [PubMed]
- Yang YX, Hu HD, Zhang DZ, et al. Identification of proteins responsible for the development of adriamycin resistance in human gastric cancer cells using comparative proteomics analysis. J Biochem Mol Biol 2007;40:853-60. [PubMed]
- Cazzaniga G, Dell’Oro MG, Mecucci C, et al. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 2005;106:1419-22. [PubMed]
- Shi SL, Li QF, Liu QR, et al. Nuclear matrix protein, prohibitin, was down-regulated and translocated from nucleus to cytoplasm during the differentiation of osteosarcoma MG-63 cells induced by ginsenoside Rg1, cinnamic acid, and tanshinone IIA (RCT). J Cell Biochem 2009;108:926-34. [PubMed]
- Sripathi SR, He W, Atkinson CL, et al. Mitochondrial-nuclear communication by prohibitin shuttling under oxidative stress. Biochemistry 2011;50:8342-51. [PubMed]
- Merkwirth C, Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta 2009;1793:27-32.
- Rastogi S, Joshi B, Fusaro G, et al. Camptothecin induces nuclear export of prohibitin preferentially in transformed cells through a CRM-1-dependent mechanism. J Biol Chem 2006;281:2951-9. [PubMed]
- Ritterson Lew C, Tolan DR. Targeting of several glycolytic enzymes using RNA interference reveals aldolase affects cancer cell proliferation through a non-glycolytic mechanism. J Biol Chem 2012;287:42554-63. [PubMed]
- Bacher S, Achatz G, Schmitz ML, et al. Prohibitin and prohibitone are contained in high-molecular weight complexes and interact with alpha-actinin and annexin A2. Biochimie 2002;84:1207-20. [PubMed]
- Pasupuleti N, Leon L, Carraway KL 3rd, et al. 5-Benzylglycinyl-amiloride kills proliferating and nonproliferating malignant glioma cells through caspase-independent necroptosis mediated by apoptosis-inducing factor. J Pharmacol Exp Ther 2013;344:600-15. [PubMed]
- Cregan SP, Dawson VL, Slack RS, et al. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004;23:2785-96. [PubMed]
- Candé C, Cohen I, Daugas E, et al. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie 2002;84:215-22. [PubMed]
- Cheng MH, Yang YC, Wong YH, et al. B1, a novel topoisomerase II inhibitor, induces apoptosis and cell cycle G1 arrest in lung adenocarcinoma A549 cells. Anticancer Drugs 2012;23:191-9. [PubMed]
- Choi YK, Seo HS, Choi HS, et al. Induction of Fas-mediated extrinsic apoptosis, p21WAF1-related G2/M cell cycle arrest and ROS generation by costunolide in estrogen receptor-negative breast cancer cells, MDA-MB-231. Mol Cell Biochem 2012;363:119-28. [PubMed]
- Smits VA, Klompmaker R, Vallenius T, et al. p21 inhibits Thr161 phosphorylation of Cdc2 to enforce the G2 DNA damage checkpoint. J Biol Chem 2000;275:30638-43. [PubMed]