

Challenges and opportunities of using stereotactic body radiotherapy with anti-angiogenesis agents in tumor therapy
Introduction
Though stereotactic body radiotherapy (SBRT), also called as stereotactic ablative radiotherapy (SABR) or stereotactic radiosurgery (SRS), was developed over 60 years ago, due to serious collateral damage to surrounding normal tissue, SBRT could not be applied extensively in clinic until the arrival of precision radiotherapy (RT) about two decades ago. Both radiation delivery and image guidance are vital technological advances that enable delivery of large doses of radiation to tumors thereby reducing damage to surrounding normal tissues. SBRT has been approved as an important treatment modality in patients with cancers of the lung, liver, spine, kidney, pancreas tumors and oligometastatic disease. Typically, SBRT involves the delivery of one or a few large dose fractions of 5 to 30 Gy per fraction (usually applied at 1 to 5 dose fractions). Variations of SBRT include a single relatively large dose RT, i.e. single fraction radiotherapy (SFRT), or more than one dose or hypo-fractioned radiotherapy (HFRT). The latter being a major paradigm (usually no less than 5.0 Gy per fraction) shift from conventional fraction radiotherapy (CFRT) which delivers several fractions of small doses ranging from 1.8 to 3.0 Gy.
While normal blood vessels aid the delivery of oxygen, nutrients and immune cells to every part of body, abnormal microvasculature within tumors play a critical role in its growth. Pro-angiogenic overwhelm anti-angiogenic factors resulting in the proliferation and migration of vascular endothelial cells (ECs) resulting in abnormalities in organization, structure and function of the tumor microvasculature. The excessive ECs and abnormal perivascular cells become disorganized and form dilated, tortuous, hyperpermeable and dysfunctional microvessels. This impedes immune cell migration and function during angiogenesis, and makes the tumor more likely to metastasize. This abnormal microvasculature with poor transportation efficiency causes hypoxia, acidosis and high interstitial fluid pressure in tumors.
Here, we review the negative effects of abnormal tumor microvasculature on SBRT and comb through literature for a better understanding of the relationship between SBRT and microvasculature in tumors, and end by discussing the combination of SBRT with anti-angiogenesis (AA) agents for cancer treatment in preclinical and clinical practice, examining the current situation of this combination therapy in anti-tumor treatment.
Microvasculature in tumor
Folkman in 1971 hypothesized that the most common pathway for new microvessel development in malignancy is angiogenesis. Under physiological conditions, pro-angiogenic factors and anti-angiogenic factors maintain a dynamic balance during the normal development of blood vessels. However, in malignant tumors, this balance is perturbed by hypoxia. Excessive pro-angiogenic factors induce abnormal growth of microvessel, which results in poor transportation efficiency and intensifying hypoxia and acidosis within the tumor microenvironment.
Hypoxia had long been identified as a direct negative factor for RT since irradiation is dependent on the existence of molecular oxygen (O2) for its cytotoxic effects. In hypoxic microenvironments, free-radicals mediate DNA damage, since O2 is necessary to produce DNA damaging free radicals produced by irradiation (1). Previous studies have proven that the linear-quadratic (LQ) model overestimates tumor cell killing by SFRT/HFRT by 20%–50% (2,3). The differential properties of SFRT/HFRT and CFRT on subpopulations of hypoxic cells are the reason for this overestimation (3). Several cellular and molecular processes caused or intensified by hypoxia, including but not limited to, induction of cancer “stem cell” phenotype, tumor growth and genomic instability, epithelial to mesenchymal transition, invasion and metastasis, have been identified to affect tumor treatment modalities when subjected to RT (4).
Microvasculature response to SFRT/HFRT/AA agents in preclinical tumor models
It remains unclear that with a similar biologically effective dose (BED) in LQ models, SFRT/HFRT kills fewer tumor cells directly compared to CFRT because of hypoxia, while SFRT/HFRT achieves better local control than CFRT (2,3). One thought is that microvessels may respond to SFRT/HFRT, which enhance the anti-tumor effect of irradiation.
In 2003, Garcia-Barros and his group discovered that high-dose SFRT (more than 8–11 Gy) facilitates apoptosis of ECs in a dose-dependent manner and normalize tumor vasculature (5). Then Fuks et al. observed that EC damage appears to be induced by both SFRT and CFRT. The different outcomes after irradiation between these two approaches may lie in the fact that EC apoptosis contribute significantly to tumor cell lethality in SFRT and EC damage induced by low-dose exposure of CFRT. This does not enhance tumor cell death effectively, as death signaling pathways in ECs are repressed by concomitant activation of tumor cell hypoxia inducible factor-1 (HIF-1) (6). A recent study came to the conclusion that using a pancreatic tumor model, irradiation with SFRT (24 Gy/1 fraction) could cause temporary vascular dysfunction and expression of HIF-1 (7). Lan et al. confirmed that SFRT/HFRT (both 12 Gy/1 fraction and 12 Gy/3 fractions) could reduce microvessel density (MVD) significantly in Lewis lung carcinoma (LLC) models (8). These results suggest that SFRT/HFRT may reduce MVD and normalize tumor microvasculature. However, more evidence is needed to explain the difference in mechanism in ECs responding to CFRT and SFRT/HFRT.
Compared to SFRT/HFRT, the mechanism of MVD reduction and microvessel normalization of AA agents in tumor is more explicit. AA agents can inhibit angiogenesis, normalize microvasculature and alleviate hypoxia by increasing vascular pericyte coverage, and reducing tumor hyperpermeability, vessel calibers, hypoxia and interstitial fluid pressure. Normalization of blood vessels occurs when there is a “window of opportunity” (4,9-11). An increase in transient perfusion during the acute phase (1 to 3 days after medicine infusion) progressively decreased tumor microvessel density on the 5th to 7th day post-AA agent therapy, resulting in decreased blood perfusion (9,12). Duration of this “window” is dependent on dose and potency of AA therapy.
In addition, microvessels may play a role in post-SFRT/HFRT tumor progression. Lan et al. observed that tumor MVD increased along with tumor regrowth after 3 weeks of a single dose of 12 Gy irradiation (8). Furthermore, irradiated tumors secrete various cytokines to inhibit apoptosis in ECs, thereby preventing vascular damage and impairing the anti-tumor effects of irradiation. HIF-1 regulates cytokines such as vascular endothelial growth factor (VEGF), which have been shown to weaken the effects of RT (13). Moreover, irradiation (6 Gy, single dose) upregulates endothelial nitric oxide synthase (eNOS) expression and activity. This activates the nitric oxide (NO) pathway in ECs. NO signaling post-irradiation induces profound transformation of the EC phenotype to generate tumor angiogenesis (14). Moreover, post-irradiation microvessels regrowth may be triggered by irradiation-induced and caspase-3-mediated cell death in both human colon cancer and human glioma xenografts (15,16).
It is still controversial whether ECs originate from bone marrow-derived cells is the key process in the revascularization of post-irradiated tumors. In certain murine and human tumors, an increase in intratumoral SDF-1/CXCR4 pathway is activated by irradiation. This recruits myeloid-derived monocytes, particularly tumor-associated macrophages, which subsequently promote tumor regrowth (17,18). Further investigations indicated that myeloid-derived monocytes, recruited post-RT in glioblastomas facilitate vasculogenesis but not angiogenesis, which subsequently results in tumor re-growth (19). However, Kozin suggested that although tumor-associated macrophages and certain subgroup of monocytes promote post-RT tumor revascularization, some ECs in growing tumors may be derived from cancer stem-like cells through trans-differentiation (20).
AA agents may be more effective for the treatment of post-HFRT relapsing tumors than HFRT-naive tumors in human tumor xenograft models (21). Various cytokines and cells have been demonstrated to play a role in regulating angiogenesis after SFRT/HFRT, which supports the use of AA agents for improved efficacy.
Effects of SFRT/HFRT/SBRT with AA agents in preclinical and clinical studies
As mentioned earlier, SFRT/HFRT kills fewer tumor cells than CFRT due to the hypoxic microenvironment in tumors. AA agents may alleviate hypoxia to improve sensitivity of tumor cells to irradiation. Combination of AA agents might be effective with SBRT where microvessel redistribution plays an important role in post-SBRT relapse. From another aspect, SBRT itself could reduce MVD and normalize microvessels in tumor. Therefore, it is important to explore whether a synergistic effect exists using a combination of SBRT and AA agents. This has been tested in various preclinical and clinical studies.
Efficiency of SFRT/HFRT with AA agents in preclinical research
Using the preclinical LLC model, VEGF was induced after exposure to RT. Pre-treatment with angiostatin or a VEGF-neutralizing antibody before HFRT (20 Gy/2 fractions) was associated with a synergistic than an additive anti-tumor effect (22,23). Endostatin, an anti-angiogenic factor, showed disparate effects on tumor growth and hypoxia between high (MCa-35) and low (MCa-4) vascularized murine mammary carcinomas. The poorly-vascularized tumor (MCa-4) responded better to endostatin therapy than the highly-vascularized tumor (MCa-35) (22-24). In a follow-up study, the team found that in MCa-4 models, a combination of HFRT (6 Gy/5 fractions) and a monoclonal antibody against vascular endothelial growth factor receptor 2 (VEGFR2; DC101) inhibited tumor growth significantly when compared to the MCa-35 models (25). In addition, hypoxia-activated chemotherapy or using a HIF-1α inhibitor enhanced the effect of SFRT (8 Gy or 10 Gy/1 fraction) in sarcoma models (26,27). Gao et al. determined that bevacizumab radiosensitizes non-small cell lung cancer (NSCLC) xenografts by inhibiting the repair of DNA double-strand break in ECs and re-normalizes the tumor microvasculature (28). Supplementing this observation, several other AA agents have been noted to enhance the effects of SFRT/HFRT (12,28-33).
Efficacy of combined SBRT with AA agents in clinical trials
Nearly 100 clinical trials evaluating the combination of CFRT and AA agents have been performed over the last decade. The majority of these were phase I or II trials with most ending with disappointing results. However, two recent phase III trials (AVAGLIO and RTOG0815) demonstrated that bevacizumab improved progression-free survival (PFS) (AVAGLIO: 11 vs. 6 months; RTOG0815: 11 vs. 7 months) but not overall survival (OS) (AVAGLIO: both 17 months; RTOG0815: both 16 months) with CFRT (60 Gy/30 fractions) in patients with glioblastoma (34,35).
These trials raise the potential for combining AA agents with SBRT in the clinic. Although a number of AA agents, mainly targeting VEGF or VEGFR, have been tested with or without chemotherapy in clinical practice, these agents were seldom combined with SBRT in phase III clinical trials.
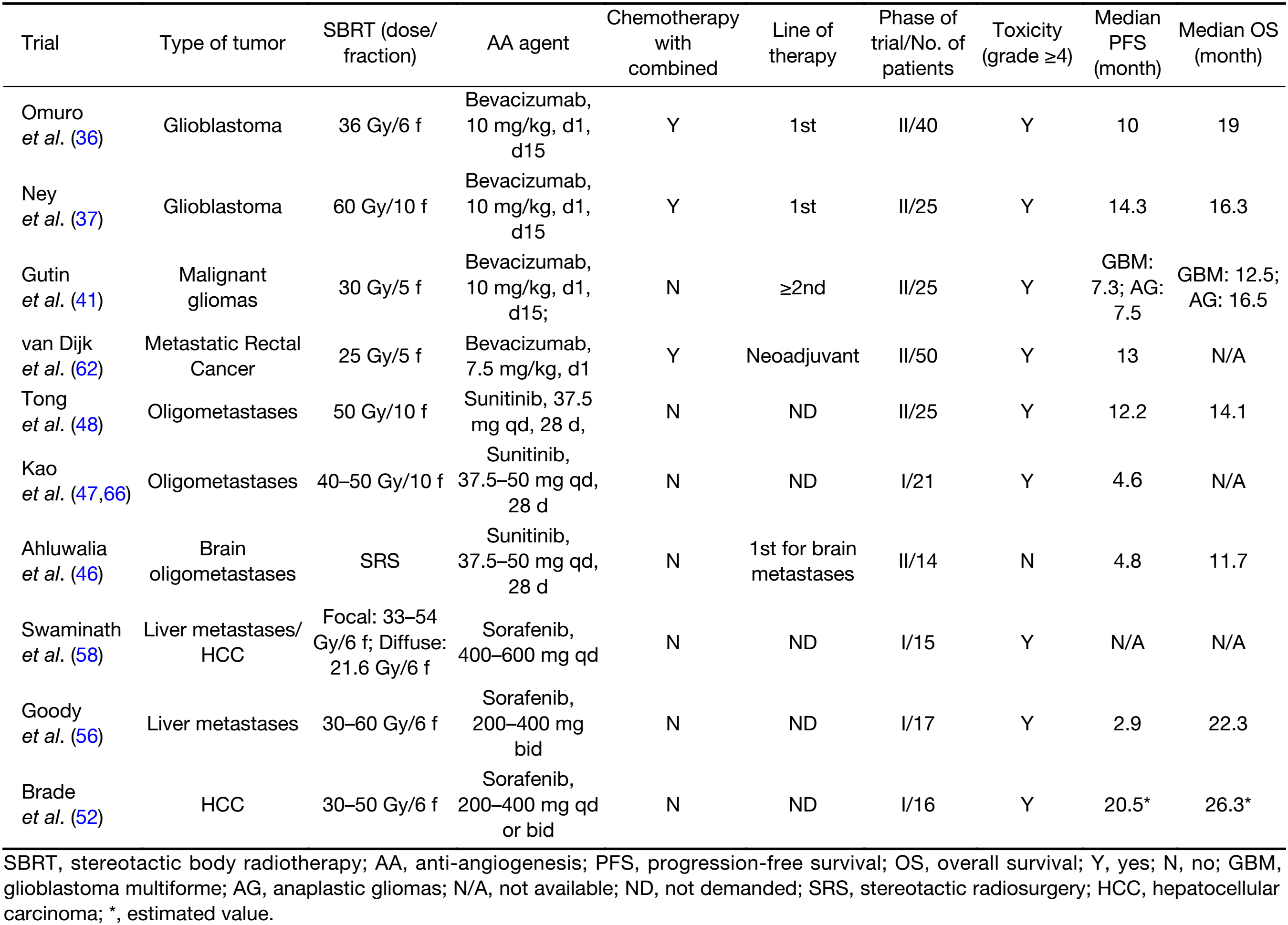
Full table
Bevacizumab together with SRS/HFRT + temozolomide, achieved a PFS of 10.0–14.3 months and an OS of 16.3–19.0 months when used as a first line of therapy against glioblastoma multiforme (GBM) (36,37). Because irradiation-induced necrosis is higher in HFRT/SRS than CFRT and bevacizumab may reduce vascular permeability, rapidly reducing peritumoral edema and alleviating subsequent radiation necrosis, bevacizumab may amplify the effect of SBS in GBM (38). Two phase II clinical trials in newly diagnosed GBM patients compared “bevacizumab + HFRT + temozolomide” and “HFRT + temozolomide” and concluded that bevacizumab in combination therapy did not prolong PFS (12.8 vs. 9.4 months, P=0.58) or OS (16.3 vs. 16.3 months) (39). The results did however indicate that bevacizumab offset the development of symptomatic radiation necrosis in small volume tumors (36), but it did not act as a radio-protective factor to these patients with large tumors after HFRT (37).
In two phase III trials (AVAGLIO and RTOG0815) in patients with newly diagnosed glioblastoma, using “CFRT (60 Gy/30 fractions) + bevacizumab + temozolomide” achieved the median PFS of 11 months for both and the median OS of 16–17 months. When the trials used HFRT/SBRT instead of CFRT (60 Gy/30 fractions), it did not make a significant difference in PFS or OS (16.3–19.0 months) (34,35).
After the initial radiation to the tumor bed, 72%–93% of GBM patients relapsed (40). The use of the combination of bevacizumab and HFRT (30 Gy/5 fractions) achieved promising outcomes in previously heavily treated GBM patients with the median tumor volume of 34 cm3: the overall response rate (ORR) was 50%, the 6-month PFS was 65%, and the median OS was 12.5 months (41). This trial provided a choice for GBM patients who relapsed. These outcomes were better than previous retrospective and prospective studies using either HFRT or CFRT without AA agents, excepting the trials that recruited patients with much smaller tumor volumes (42). In a single-institution retrospective study of high-grade glioma patients treated with CFRT (36 Gy/18 fractions) with concurrent bevacizumab, the OS (12.1months) appeared to be similar when compared with those who received HFRT with bevacizumab, better than that of CFRT alone or CFRT with concurrent temozolomide (8.0 months) (43). However, this retrospective study did not provide the target tumor volume, which is a recognized prognostic factor. Additional studies are needed to confirm this.
Oligometastasis (defined as five or less clinically detectable metastatic lesions from any primary site) signifies a limited tumor burden that is potentially curable with appropriate local treatment. The use of SBRT could benefit patients with liver or lung oligometastatic disease (44,45). Sunitinib in combination with SBRT has been used to treat patients with oligometastasis of head and neck, liver, lung, kidney and prostate tumors. Sunitinib combined with SRS to treat intracranial oligometastasis resulted in shorter median PFS than with SBRT to the extracranial lesions (4.8 vs. 12.2 months) (46-48). On the other hand, when compared with the results of a phase Ib trial for brain CFRT + sunitinib, the median PFS was 5 months, and the median OS was 8.8 months (49). We could infer that using SBRT or CFRT to brain did not make a difference in combination with sunitinib. Presumably, these combination trials involved different types of tumors, since sunitinib, as a single agent, was ineffective at controlling extracranial disease. The small sample size and possible biases may have led to misinterpretation of the results. In fact, patients with a large tumor burden were associated with worse survival prognosis when treated with SBRT alone. Patients with kidney or prostate primary carcinoma suggested improved OS when treated with SBRT and sunitinib (47,50).
Since sorafenib alone has been verified to improve the PFS and OS of patients with hepatocellular carcinoma (HCC) (51), it has been hypothesized that combining sorafenib and SBRT may increase the efficacy in patients with HCC or liver metastases. A phase I clinical trial, using “SBRT + sorafenib” in HCC patients, reported prematurely because of unacceptable toxicities with an estimated median PFS (12.1 months) and estimated median OS (26.3 months) (52). However, earlier phase I and II trials carried out using SBRT alone in HCC patients had a median PFS of 6.0 months and a median OS of 17.0 months (53). Another phase II clinical trial that used CFRT (40–60 Gy/20–30 fractions) with sorafenib in advanced HCC patients resulted in an ORR of 55%, a median PFS of 8.6 months and a median OS of 14 months (54). Two other clinical trials in patients undergoing SBRT with or without sorafenib for liver metastases indicated no differences in ORR (47% vs. 49%) nor median OS (20.9 vs. 17.9 months) (55,56).
Toxicities in clinical practices
In general, SBRT with AA agents had a moderate effect for certain tumors, such as heavily treated GBM, renal cell carcinoma and prostate carcinoma (41,47). However, grade 4 toxicities were seldom observed in patients receiving AA agents with CFRT, while the combination of AA agents and SBRT had much higher rates of serious toxicities (56-58). These toxicities were also observed in SBRT/SRS/AA agents alone or in combination therapy. These severe and relatively high-frequency toxicities are particularly gastrointestinal and hepatic toxicities in combination of SBRT with AA agents. Additional studies are needed to determine whether any particular agent is safer to use with SBRT than others.
Large doses of irradiation could cause damage to normal adjacent tissue. This usually results in severe inflammation, pain, dysfunction, radioactive gastritis, esophagitis, pneumonitis, liver dysfunction and intracranial radiation necrosis (45,55,59). Radioactive pneumonitis may progress to pulmonary fibrosis without effective anti-inflammatory treatment. Radionecrosis is due to SRS and usually occurs 6 months after irradiation. In a clinical trial using SBS + bevacizumab in GBM patients with large tumor volumes [median planning target volume 1 (PTV1) of 131 cm], there was an unexpected high risk of radionecrosis, which developed in 30 out of 35 patients. However, the incidence of radionecrosis in patients receiving CFRT + bevacizumab has not been reported for the two phase III trials (34,35).
AA agent-associated toxicities are numerous and mainly consist of cardiovascular and non-cardiovascular toxicities, which depend on the category of AA agents used. Hypertension and thromboembolic diseases are common cardiovascular toxicities induced by AA agents, while others include myocardial ischemia, left ventricular dysfunction and prolongation of the QT interval. Non-cardiovascular adverse effects include proteinuria, gastrointestinal toxicities, delayed wound healing, fatigue, stomatitis, thyroid dysfunction, myelosuppression, dysphonia and cutaneous effects. Additional rare or agent-specific toxicities include reversible posterior leukoencephalopathy, microangiopathic hemolysis, osteonecrosis of the jaw, hypoglycemia and pancreatic enzyme elevations.
Bevacizumab induces more gastrointestinal toxicity than other AA agents (60). This may be due to bevacizumab’s ability to form immune complexes and induce vascular inflammation and clotting. Gastrointestinal toxicities are significantly reduced with a multi-fraction regimen (25–45 Gy in 3–5 fractions with median BED3 of 105.6 Gy; range, 66.7–180.0 Gy) compared with a single fraction (25 Gy in 1 fraction or BED3 of 233.3 Gy) for SBRT in pancreatic tumor (61). Thus, adopting HFRT in addition to SFRT, and using the lowest BED, which provides high local control, and establishing an optimal dose should be investigated for concurrent or possible sequential AA therapy.
The most common grade ≥3 non-surgery-associated toxicities are diarrhea (12%, 6/50), pulmonary embolisms (12%, 6/50) when using “bevacizumab + capecitabine + oxaliplatin + HFRT (5 Gy/5 fractions)” as a neoadjuvant scheme for metastasized rectal carcinoma (62). However, when HFRT is replaced by CFRT in this neoadjuvant scheme, grade ≥3 toxicities were substantially reduced with only 2 of 55 patients developing deep vein thrombosis but having similar rates of diarrhea (12.7%, 7/55) (63). Lymphopenia (36%, 9/25), thrombocytopenia (12%, 3/25) and hyponatremia (24%, 6/25) were common toxicities for combination of bevacizumab + HFRT (6 Gy/5 fractions) for recurrent gliomas (41). A retrospective study found that esophageal fistula was a rare complication (5.8%, 3/52) in patients with irradiated volume within 2 cm of the esophagus. The fistulas occurred only when doses exceed 51 Gy and were only observed in the patients treated with AA agents post SBRT (64). Most of these hematopoietic toxicities were reversed by effective medication. Other rare toxicities observed were renal failure, wound healing complications, infections, ischemic stroke and central nervous system bleeding.
Sunitinib seems to be associated with less serious toxicities than bevacizumab. Patients with oligometastasis irradiated with SBRT experienced grade ≥3 toxicities including anemia (5.4%, 3/56), neutropenia (10.7%–14.3%, 6/56 and 2/14), thrombocytopenia (12.5%, 7/56), bleeding (5.4%, 3/56), liver function test abnormality (5.4%, 3/56), metabolic abnormality (3.6%, 2/56), fatigue (35.7%, 5/14), lymphopenia, rash, mucositis and hemolysis (7.1%, 1/14) (46,47).
Sorafenib with SBRT, used in hepatic metastasis and HCC patients, manifested the unexpected rapid reduction of liver volume (52,58). It might necessitate individualized assessments for probable liver volume reduction prior to SBRT in patients taking sorafenib. Significant gastrointestinal toxicities experienced with this combination may be attributed to gastrointestinal structures receiving larger doses of irradiation than planned because of liver deformation (58,65). However, combination of sorafenib with CFRT revealed acceptable toxicities, with 7.5% (3/40) of patients experiencing diarrea and 5.0% (2/40) suffering from impaired liver function, which subsequently lead to sorafenib reduction or discontinuation (54).
It is undetermined whether a “wash-out” period should be observed between AA therapy and SBRT. Presumably, a “wash-out” period after radiation is dependent on the half-life of the specific AA agent. The optimal timing, duration and dosing of AA agents when combined with SBRT also remain unknown. It is however not recommended that concurrent sorafenib with SBRT be a strategy for further clinical testing in patients with locally advanced HCC, particularly with irradiation of a large area of the liver (52). Sequential SBRT plus sorafenib (half dose for the first 4 weeks) in RTOG 1112 in locally advanced HCC is a strategy being currently investigated. Table 1 lists the outcomes of completed clinical trials testing the combination of AA agents with SBRT.
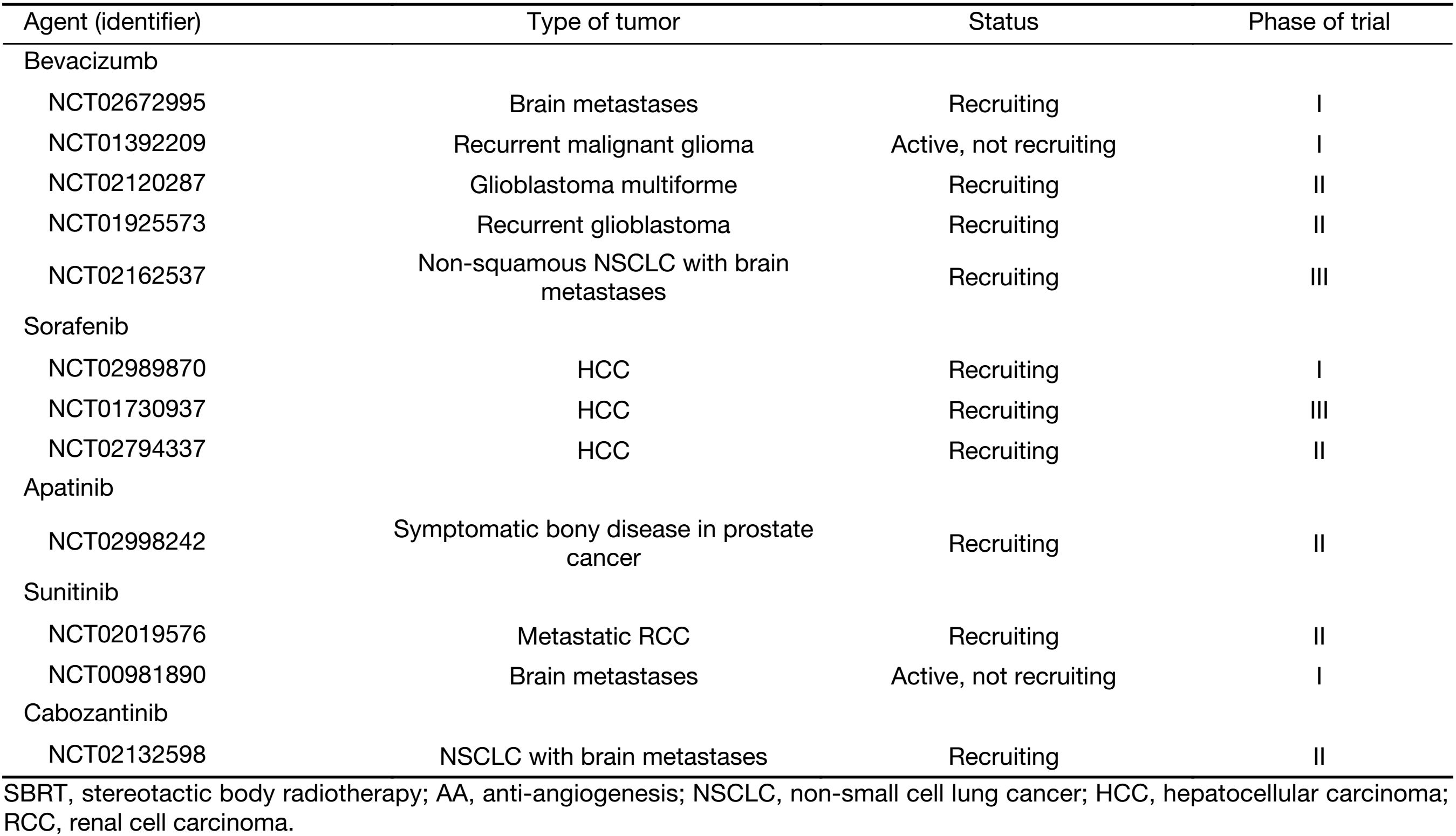
Results of a search in “clinicaltrials.gov” using the phrases, “stereotactic body radiation therapy” or “stereotactic ablative radiotherapy” or “stereotactic radiosurgery” for completed, not completed or terminated clinical trials involving both SBRT and AA agents are listed in Table 2. However, it is too early to conclude whether AA agents synergize the cytotoxicity of SBRT.
Full table
Summary and perspective
More compact evidence is needed to confirm and optimize the combination of SBRT and AA therapy, as well as to determine optimal timing, dose and washout periods. A number of clinical trials are underway to answer these questions. Although preclinical models have indicated that SBRT with AA agents has a synergistic effect on efficacy, the treatment tends to induce more adverse events compared to CFRT with AA agents. Due to the small number of patients with successful outcomes, subsequent clinical trials may need to be more selective when recruiting patients. We have listed some of the reasons why combined therapy has not achieved greater success in clinical practice to date as follows:
Firstly, only limited RT clinical trials and even fewer SBRT trials have been conducted. Of the 200 clinical trials supported by the American National Cancer Institute (NCI), only 11 involved RT (67). Nevertheless, brighter prospects lie ahead as a new era dawned with hypofractionation in general, and SBRT in particular, promising to give RT an edge over other cancer therapies.
Secondly, experimental designs and animal models need to be improved. In general, tumors in animal models are simple at genetic level and grow so rapidly that they tend to be more sensitive to AA agents than corresponding tumors in humans. Improvement of patient-derived xenograft models depends on the evolution of more humanized immune system in mouse. Thus, the findings with preclinical models may not corroborate with treatment setting in clinical practices (4).
Thirdly, safety should be given particular attention. Combined treatment may cause significant side effects, including prominent gastrointestinal or hepatic toxicities and other severe potential adverse reactions.
In the near future, more clinical trials on SBRT and AA combination therapy will reveal the benefit to patients with certain types of cancer. In addition, personalized medicine is warranted since tumor heterogeneity within a patient can lead to varied responses to combination therapy.
Acknowledgements
This study was supported by National Natural Science Foundation of China (No. 81672982, 81602670) and Sichuan Province Research Foundation for Basic Research (No. 2016JY0050).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 2004;4:437–47. [PubMed] DOI:10.1038/nrc1367
- Brown JM, Koong AC. High-dose single-fraction radiotherapy: exploiting a new biology? Int J Radiat Oncol Biolo Phys 2008;71:324–5. [PubMed] DOI:10.1016/j.ijrobp.2008.02.003
- Carlson DJ, Keall PJ, Loo BW Jr., et al. Hypofractionation results in reduced tumor cell kill compared to conventional fractionation for tumors with regions of hypoxia. Int J Radiat Oncol Biol Phys 2011;79:1188–95. [PubMed] DOI:10.1016/j.ijrobp.2010.10.007
- Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 2014;26:605–22. [PubMed] DOI:10.1016/j.ccell.2014.10.006
- Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003;300:1155–9. [PubMed] DOI:10.1126/science.1082504
- Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell 2005;8:89–91. [PubMed] DOI:10.1016/j.ccr.2005.07.014
- Maeda A, Chen Y, Bu J, et al. In vivo imaging reveals significant tumor vascular dysfunction and increased tumor hypoxia-inducible factor-1α expression induced by high single-dose irradiation in a pancreatic tumor model. Int J Radiat Oncol Biol Phys 2017;97:184–94. [PubMed] DOI:10.1016/j.ijrobp.2016.09.005
- Lan J, Wan XL, Deng L, et al. Ablative hypofractionated radiotherapy normalizes tumor vasculature in lewis lung carcinoma mice model. Radiat Res 2013;179:458–64. [PubMed] DOI:10.1667/RR3116.1
- Dickson PV, Hamner JB, Sims TL, et al. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res 2007;13:3942–50. [PubMed] DOI:10.1158/1078-0432.CCR-07-0278
- Franco M, Man S, Chen L, et al. Targeted anti-vascular endothelial growth factor receptor-2 therapy leads to short-term and long-term impairment of vascular function and increase in tumor hypoxia. Cancer Res 2006;66:3639–48. [PubMed] DOI:10.1158/0008-5472.CAN-05-3295
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011;10:417–27. [PubMed] DOI:10.1038/nrd3455
- Meng MB, Jiang XD, Deng L, et al. Enhanced radioresponse with a novel recombinant human endostatin protein via tumor vasculature remodeling: experimental and clinical evidence. Radiother Oncol 2013;106:130–7. [PubMed] DOI:10.1016/j.radonc.2012.10.022
- Moeller BJ, Cao Y, Li CY, et al. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004;5:429–41. [PubMed] DOI:10.1016/S1535-6108(04)00115-1
- Sonveaux P, Brouet A, Havaux X, et al. Irradiation-induced angiogenesis through the up-regulation of the nitric oxide pathway: implications for tumor radiotherapy. Cancer Res 2003;63:1012–9. [PubMed]
- Feng X, Tian L, Zhang Z, et al. Caspase 3 in dying tumor cells mediates post-irradiation angiogenesis. Oncotarget 2015;6:32353–67. [PubMed] DOI:10.18632/oncotarget.5898
- Feng X, Yu Y, He S, et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett 2017;385:12–20. [PubMed] DOI:10.1016/j.canlet.2016.10.042
- Kozin SV, Kamoun WS, Huang Y, et al. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res 2010;70:5679–85. [PubMed] DOI:10.1158/0008-5472.CAN-09-4446
- Chen FH, Fu SY, Yang YC, et al. Combination of vessel-targeting agents and fractionated radiation therapy: the role of the SDF-1/CXCR4 pathway. Int J Radiat Oncol Biol Phys 2013;86:777–84. [PubMed] DOI:10.1016/j.ijrobp.2013.02.036
- Kioi M, Vogel H, Schultz G, et al. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 2010;120:694–705. [PubMed] DOI:10.1172/JCI40283
- Kozin SV, Duda DG, Munn LL, et al. Is vasculogenesis crucial for the regrowth of irradiated tumours?. Nat Rev Cancer 2011;11:532. [PubMed] DOI:10.1038/nrc2007-c1
- Kozin SV, Winkler F, Garkavtsev I, et al. Human tumor xenografts recurring after radiotherapy are more sensitive to anti-vascular endothelial growth factor receptor-2 treatment than treatment-naive tumors. Cancer Res 2007;67:5076–82. [PubMed] DOI:10.1158/0008-5472.CAN-06-3664
- Mauceri HJ, Hanna NN, Beckett MA, et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature 1998;394:287–91. [PubMed] DOI:10.1038/28412
- Gorski DH, Beckett MA, Jaskowiak NT, et al. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res 1999;59:3374–8. [PubMed]
- Fenton BM, Paoni SF, Grimwood BG, et al. Disparate effects of endostatin on tumor vascular perfusion and hypoxia in two murine mammary carcinomas. Int J Radiat Oncol Biol Phys 2003;57:1038–46. [PubMed] DOI:10.1158/0008-5472.CAN-04-0434
- Fenton BM, Paoni SF, Ding I. Pathophysiological effects of vascular endothelial growth factor receptor-2-blocking antibody plus fractionated radiotherapy on murine mammary tumors. Cancer Res 2004;64:5712–9. [PubMed] DOI:10.1158/0008-5472.CAN-04-0434
- Lee HJ, Yoon C, Park DJ, et al. Inhibition of vascular endothelial growth factor A and hypoxia-inducible factor 1α maximizes the effects of radiation in sarcoma mouse models through destruction of tumor vasculature. Int J Radiat Oncol Biol Phys 2015;91:621–30. [PubMed] DOI:10.1016/j.ijrobp.2014.10.047
- Yoon C, Lee HJ, Park DJ, et al. Hypoxia-activated chemotherapeutic TH-302 enhances the effects of VEGF-A inhibition and radiation on sarcomas. Br J Cancer 2015;113:46–56. [PubMed] DOI:10.1038/bjc.2015.186
- Gao H, Xue J, Zhou L, et al. Bevacizumab radiosensitizes non-small cell lung cancer xenografts by inhibiting DNA double-strand break repair in endothelial cells. Cancer Lett 2015;365:79–88. [PubMed] DOI:10.1016/j.canlet.2015.05.011
- Cao C, Albert JM, Geng L, et al. Vascular endothelial growth factor tyrosine kinase inhibitor AZD2171 and fractionated radiotherapy in mouse models of lung cancer. Cancer Res 2006;66:11409–15. [PubMed] DOI:10.1158/0008-5472.CAN-06-2414
- Cuneo KC, Geng L, Fu A, et al. SU11248 (sunitinib) sensitizes pancreatic cancer to the cytotoxic effects of ionizing radiation. Int J Radiat Oncol Biol Phys 2008;71:873–9. [PubMed] DOI:10.1016/j.ijrobp.2008.02.062
- Landuyt W, Ahmed B, Nuyts S, et al. In vivo antitumor effect of vascular targeting combined with either ionizing radiation or anti-angiogenesis treatment. Int J Radiat Oncol Biol Phys 2001;49:443–50. [PubMed] DOI:10.1016/S0360-3016(00)01470-X
- Rao SS, Thompson C, Cheng J, et al. Axitinib sensitization of high Single Dose Radiotherapy. Radiother Oncol 2014;111:88–93. [PubMed] DOI:10.1016/j.radonc.2014.02.010
- Riesterer O. Ionizing radiation antagonizes tumor hypoxia induced by antiangiogenic treatment. Clin Cancer Res 2006;12:3518–24. [PubMed] DOI:10.1158/1078-0432.CCR-05-2816
- Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 2014;370:699–708. [PubMed] DOI:10.1056/NEJMoa1308573
- Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 2014;370:709–22. [PubMed] DOI:10.1056/NEJMoa1308345
- Omuro A, Beal K, Gutin P, et al. Phase II study of bevacizumab, temozolomide, and hypofractionated stereotactic radiotherapy for newly diagnosed glioblastoma. Clin Cancer Res 2014;20:5023–31. [PubMed] DOI:10.1158/1078-0432.CCR-14-0822
- Ney DE, Carlson JA, Damek DM, et al. Phase II trial of hypofractionated intensity-modulated radiation therapy combined with temozolomide and bevacizumab for patients with newly diagnosed glioblastoma. J Neurooncol 2015;122:135–43. [PubMed] DOI:10.1007/s11060-014-1691-z
- Levin VA, Bidaut L, Hou P, et al. Randomized double-blind placebo-controlled trial of bevacizumab therapy for radiation necrosis of the central nervous system. Int J Radiat Oncol Biol Phys 2011;79:1487–95. [PubMed] DOI:10.1016/j.ijrobp.2009.12.061
- Carlson JA, Reddy K, Gaspar LE, et al. Hypofractionated-intensity modulated radiotherapy (hypo-IMRT) and temozolomide (TMZ) with or without bevacizumab (BEV) for newly diagnosed glioblastoma multiforme (GBM): a comparison of two prospective phase II trials. J Neurooncol 2015;123:251–7. [PubMed] DOI:10.1007/s11060-015-1791-4
- Buglione M, Pedretti S, Poliani PL, et al. Pattern of relapse of glioblastoma multiforme treated with radical radio-chemotherapy: Could a margin reduction be proposed? J Neurooncol 2016;128:303–12. [PubMed] DOI:10.1007/s11060-016-2112-2
- Gutin PH, Iwamoto FM, Beal K, et al. Safety and efficacy of bevacizumab with hypofractionated stereotactic irradiation for recurrent malignant gliomas. Int J Radiat Oncol Biol Phys 2009;75:156–63. [PubMed] DOI:10.1016/j.ijrobp.2008.10.043
- Ryu S, Buatti JM, Morris A, et al. The role of radiotherapy in the management of progressive glioblastoma: a systematic review and evidence-based clinical practice guideline. J Neurooncol 2014;118:489–99. [PubMed] DOI:10.1007/s11060-013-1337-6
- Niyazi M, Ganswindt U, Schwarz SB, et al. Irradiation and bevacizumab in high-grade glioma retreatment settings. Int J Radiat Oncol Biol Phys 2012;82:67–76. [PubMed] DOI:10.1016/j.ijrobp.2010.09.002
- Rusthoven KE, Kavanagh BD, Cardenes H, et al. Multi-institutional phase I/II trial of stereotactic body radiation therapy for liver metastases. J Clin Oncol 2009;27:1572–8. [PubMed] DOI:10.1200/JCO.2008.19.6329
- Rusthoven KE, Kavanagh BD, Burri SH, et al. Multi-institutional phase I/II trial of stereotactic body radiation therapy for lung metastases. J Clin Oncol 2009;27:1579–84. [PubMed] DOI:10.1200/JCO.2008.19.6386
- Ahluwalia MS, Chao ST, Parsons MW, et al. Phase II trial of sunitinib as adjuvant therapy after stereotactic radiosurgery in patients with 1-3 newly diagnosed brain metastases. J Neurooncol 2015;124:485–91. [PubMed] DOI:10.1007/s11060-015-1862-6
- Kao J, Chen CT, Tong CC, et al. Concurrent sunitinib and stereotactic body radiotherapy for patients with oligometastases: final report of a prospective clinical trial. Target Oncol 2014;9:145–53. [PubMed] DOI:10.1007/s11523-013-0280-y
- Tong CC, Ko EC, Sung MW, et al. Phase II trial of concurrent sunitinib and image-guided radiotherapy for oligometastases. PLoS One 2012;7:e36979. [PubMed] DOI:10.1371/journal.pone.0036979
- Wuthrick EJ, Kamrava M, Curran WJ Jr., et al. A phase 1b trial of the combination of the antiangiogenic agent sunitinib and radiation therapy for patients with primary and metastatic central nervous system malignancies. Cancer 2011;117:5548–59. [PubMed] DOI:10.1002/cncr.26216
- Milano MT, Katz AW, Zhang H, et al. Oligometastases treated with stereotactic body radiotherapy: long-term follow-up of prospective study. Int J Radiat Oncol Biol Phys 2012;83:878–86. [PubMed] DOI:10.1016/j.ijrobp.2011.08.036
- Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378–90. [PubMed] DOI:10.1056/NEJMoa0708857
- Brade AM, Ng S, Brierley J, et al. Phase 1 trial of sorafenib and stereotactic body radiation therapy for hepatocellular carcinoma. Int J Radiat Oncol Biol Phys 2016;94:580–7. [PubMed] DOI:10.1016/j.ijrobp.2015.11.048
- Bujold A, Massey CA, Kim JJ, et al. Sequential phase I and II trials of stereotactic body radiotherapy for locally advanced hepatocellular carcinoma. J Clin Oncol 2013;31:1631–9. [PubMed] DOI:10.1200/JCO.2012.44.1659
- Chen SW, Lin LC, Kuo YC, et al. Phase 2 study of combined sorafenib and radiation therapy in patients with advanced hepatocellular carcinoma. Int J Radiat Oncol Biol Phys 2014;88:1041–7. [PubMed] DOI:10.1016/j.ijrobp.2014.01.017
- Lee MT, Kim JJ, Dinniwell R, et al. Phase I study of individualized stereotactic body radiotherapy of liver metastases. J Clin Oncol 2009;27:1585–91. [PubMed] DOI:10.1200/JCO.2008.20.0600
- Goody RB, Brade AM, Wang L, et al. Phase I trial of radiation therapy and sorafenib in unresectable liver metastases. Radiother Oncol 2017;123:234–39. [PubMed] DOI:10.1016/j.radonc.2017.01.018
- Pollom EL, Deng L, Pai RK, et al. Gastrointestinal toxicities with combined antiangiogenic and stereotactic body radiation therapy. Int J Radiat Oncol Biol Phys 2015;92:568–76. [PubMed] DOI:10.1016/j.ijrobp.2015.02.016
- Swaminath A, Knox JJ, Brierley JD, et al. Changes in liver volume observed following sorafenib and liver radiation therapy. Int J Radiat Oncol Biol Phys 2016;94:729–37. [PubMed] DOI:10.1016/j.ijrobp.2015.12.015
- Salama JK, Hasselle MD, Chmura SJ, et al. Stereotactic body radiotherapy for multisite extracranial oligometastases: final report of a dose escalation trial in patients with 1 to 5 sites of metastatic disease. Cancer 2012;118:2962–70. [PubMed] DOI:10.1002/cncr.26611
- Barney BM, Markovic SN, Laack NN, et al. Increased bowel toxicity in patients treated with a vascular endothelial growth factor inhibitor (VEGFI) after stereotactic body radiation therapy (SBRT). Int J Radiat Oncol Biol Phys 2013;87:73–80. [PubMed] DOI:10.1016/j.ijrobp.2013.05.012
- Pollom EL, Alagappan M, von Eyben R, et al. Single- versus multifraction stereotactic body radiation therapy for pancreatic adenocarcinoma: outcomes and toxicity. Int J Radiat Oncol Biol Phys 2014;90:918–25. [PubMed] DOI:10.1016/j.ijrobp.2014.06.066
- van Dijk TH, Tamas K, Beukema JC, et al. Evaluation of short-course radiotherapy followed by neoadjuvant bevacizumab, capecitabine, and oxaliplatin and subsequent radical surgical treatment in primary stage IV rectal cancer. Ann Oncol 2013;24:1762–9. [PubMed] DOI:10.1093/annonc/mdt124
- Landry JC, Feng Y, Cohen SJ, et al. Phase 2 study of preoperative radiation with concurrent capecitabine, oxaliplatin, and bevacizumab followed by surgery and postoperative 5-fluorouracil, leucovorin, oxaliplatin (FOLFOX), and bevacizumab in patients with locally advanced rectal cancer: ECOG 3204. Cancer 2013;119:1521–7. [PubMed] DOI:10.1002/cncr.27890
- Stephans KL, Diaconu CI, Djemil T, et al. Stereotactic radiation near the esophagus: when does a fistula occur? J Thorac Oncol 2012;7:S223–S224. DOI:10.1097/JTO.0b013e318269fc07
- Chen J, Bissonnette JP, Craig T, et al. Investigation into delivered doses for hepatocellular carcinoma patients treated with concurrent sorafenib and stereotactic-body radiotherapy (SBRT). J Med Imaging Radiat Sci 2017;48:S14. DOI:10.1016/j.jmir.2017.02.043
- Kao J, Packer S, Vu HL, et al. Phase 1 study of concurrent sunitinib and image-guided radiotherapy followed by maintenance sunitinib for patients with oligometastases: acute toxicity and preliminary response. Cancer 2009;115: 3571-80.
- Brown JM, Adler JR Jr. Is equipment development stifling innovation in radiation oncology? Int J Radiat Oncol Biol Phys 2015;92:713–4. [PubMed] DOI:10.1016/j.ijrobp.2015.03.005