

Feasibility of personalized treatment concepts in gastrointestinal malignancies: Sub-group results of prospective clinical phase II trial EXACT
Introduction
Precision medicine utilizes molecular information to specify an individual disease and provide potential tailored treatment options (1). Progress in high-throughput technologies has led to the identification of multiple genomic alterations. In combination with the continuously development of new target therapies, precision medicine becomes compatible with the timeframe of clinical practice (2). Many genetic molecular alterations exist across different tumor types and histologies, thereby shifting the existing drug development strategies more and more towards a histology-agnostic molecularly based treatment (3). Some specific molecules have already proven their impact on cancer treatment considerations. This is best exemplified by the success of imatinib in chronic myelogenous leukemia (4). Since then, a number of early phase clinical trials tested targeted treatment in specific molecularly characterized subsets of patients (5-10). Those basket trials showed variable results and encouraged translational researchers to assess predictive biomarkers to determine outcome and efficacy. In gastric cancer, trastuzumab is a Food and Drug Administration (FDA) approved targeted therapy for which a biomarker of response (HER2 amplification) is available (11). Other biomarkers influence anti-epidermal growth factor receptor (EGFR) therapy (RAS mutation) (12) or determine prognostic value (BRAF V600E mutation) (13). However, prospective randomized clinical trials are lacking which clarify whether matching actionable mutations with targeted therapy will contribute to improving survival in gastrointestinal (GI) cancer patients.
Here, we have examined the concept of precision medicine in GI malignancies by using molecular profiling of patients’ tumors in the era of molecularly targeted treatments as well as immunotherapies. We have demonstrated that in a majority of patients an individualized molecular targeted treatment, based on real-time assessment of the tumor’s molecular profile (MP), might reflect an efficient strategy to control the underlying disease and exceed the efficacy of the immediately preceding standard treatment administered.
Materials and methods
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Ethic Committee of the Medical University of Vienna (Nr.1541/2012). The study is registered at Clinicaltrials.gov (No. NCT02999750).
Study design
The study (Extended Analysis for Cancer Treatment-EXACT) was defined to prospectively validate treatment benefit of an individualized treatment concept based on the MP from paraffin-embedded tumor tissue sections obtained before the start of treatment (real time biopsy). Here we aimed to evaluate the GI subgroup of the trial. The primary objective was to determine whether the individualized treatment concept resulted in a longer progression-free survival (PFS1) when compared with the last standard treatment given before (PFS0). Secondary endpoints consisted of assessment of overall treatment response rate (ORR) and overall survival (OS) by Response Evaluation Criteria in Solid Tumors (RECIST) criteria.
Patients
Patient eligibility criteria included informed consent, any histologic type of GI cancer without further standard treatment options according to international treatment guidelines, tumor progression upon treatment by RECIST criteria, age ≥18 years old, and Eastern Cooperative Oncology Group (ECOG) performance status 0−1. A fresh tumor biopsy was obtained for pathologic analysis. Biopsies were performed by interventional radiological techniques. Patients were considered to be eligible for inclusion in the study if treatment could be initiated based upon the MP derived from cancers by real-time biopsy.
Individual treatment suggestions were derived from a molecular oncology tumor board. This multidisciplinary team (MDT) meeting was held on a biweekly schedule. Results of the molecular characteristics were discussed and therapy was offered related to toxicity profiles, data derived from prospective clinical trials as well as respective patient history.
Cancer gene panel sequencing
The DNA library was generated by multiplex polymerase chain reaction (PCR) with the Ion AmpliSeq Cancer Hotspot Panel v2TM (Life Technologies, Carlsbad, CA, USA). The panel covers mutation hotspots of 50 genes, mostly oncogenes and tumor suppressor genes that are frequently mutated in tumors (ABL, AKT, ALK, APC, ATM, BRAF, CDH, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, JAK2, JAK3, IDH2, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL). Sequencing was performed with an Ion Torrent PGMTM (Life Technologies). Ambiguous nonsynonymous mutations detected with the Ion Torrent PGMTM were verified by capillary sequencing. The sequencing of PCR products was carried out with the BigDyeR Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Waltham, MA, USA). The resulting DNA fragments were purified with the DyeEx 96 Kit (QIAGEN GmbH, Hilden, Germany) and sequenced with a 3500 Genetic Analyzer (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). For sequence analysis we employed the SeqScape Version 2.7 software (Applied Biosystems).
Immunohistochemistry (IHC)
IHC was performed with a Ventana Benchmark Ultra stainer (Ventana, Tucson, AZ, USA). The following antibodies were employed: ALK (clone 1A4; Zytomed, Berlin, Germany), CD30 (clone BerH2; Dako, Vienna, Austria), CD20 (clone L26; Dako), EGFR (clone 3C6; Ventana), Estrogen-receptor (clone SP1; Ventana), HER2 (clone 4B5; Ventana), HER3 (clone SP71; Abcam), KIT (clone 9.7; Ventana), MET (clone SP44; Ventana), phospho-mTOR (clone 49F9; Cell Signalling Technology Inc., Danvers, MA, USA), PDGFRA (rabbit polyclonal; Thermo Fisher Scientific), PDGFRB (clone 28E1, Cell Signalling), PD-L1 (clone E1L3N; Cell Signalling), Progesterone-receptor (clone 1E2; Ventana), PTEN (clone Y184; Abcam) and ROS1 (clone D4D6; Cell Signalling).
Statistical analysis
For the statistical analysis on a difference between PFS0 and PFS1, the Wilcoxon Signed Rank Test was used. To find possible patient characteristics which could potentially influence the difference between PFS0 and PFS1, a linear model was performed with the difference (PFS1−PFS0) as dependent variables, and age as well as gender as independent variables. Furthermore, a 95% confidence interval (95% CI) for the median of the ratio PFS1/PFS0 was calculated through bootstrap with 1,000 samples. It has to be noted that three of the 55 patients had still an ongoing therapy at the time of the present analysis. Analysis was performed using statistical Software R (Version 3.3.0; R Foundation for Statistical Computing, Vienna, Austria).
Results
Patient characteristics and study algorithm
Patients with GI tumors after failure of standard treatment were real-time biopsied. Fresh tumor material was investigated for possible druggable targets via next generation sequencing (NGS), IHC and cytogenetic analysis. Results of the MP were discussed by a MDT within the frame of a tumor board for treatment decision (14). Out of 114 screened patients for the original EXACT trial, 55 (48%) were eligible to start targeted treatment based upon the MP derived from real-time biopsy. From those 55 patients, 21 (38%) were diagnosed with GI malignancies. Fourteen (67.0%) men and 7 (33.0%) women with a mean age of 57 (range, 26−72) years were enrolled. At time of censoring (12/31/2016), 12 (57.0%) patients were alive, while 9 (43.0%) patients were deceased. Most frequent malignancies that were enrolled and treated were colon cancer (n=7, 33.3%) and cholangiocarcinoma (CCC) (n=6, 28.6%) (Table 1).

Full table
MP
Tumor samples were analyzed via NGS, IHC and cytogenetic analysis using the MONDTI platform (14). Most frequent somatic mutations discovered were TP53 (29%), KRAS (24%), IDH1 (10%), PTEN (10%) and SMAD4 (10%) (Figure 1). IHC could reveal protein overexpression for EGFR in 86%, MET in 81%, mTOR in 76% and PDGFRa in 52% of patients. The results of cytogenetic analysis (fluorescence in situ hybridization, FISH) did not affect the treatment suggestion (data not shown).

Targeted therapy
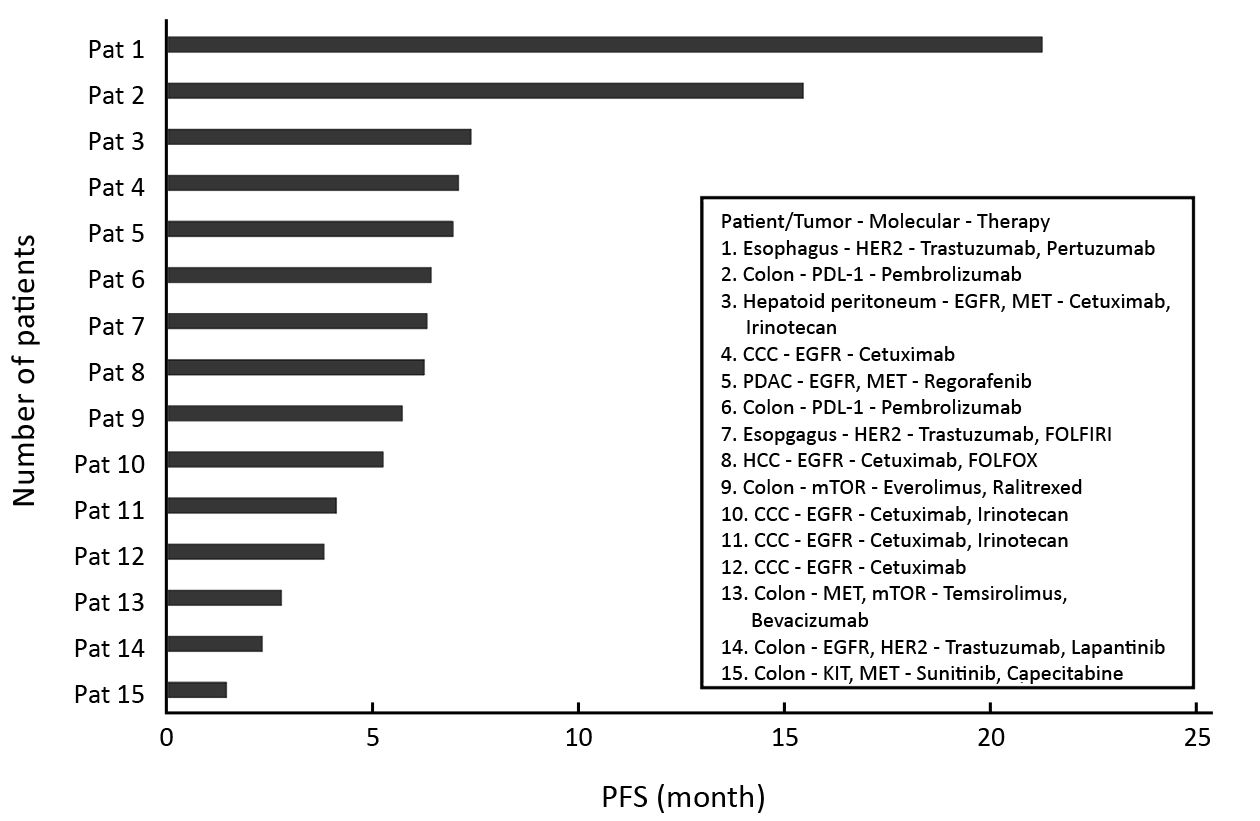
The median PFS1/PFS0 ratio of all patients was 1.59 (quartile 0.43/12.76). Out of 21 patients, 15 (71%) showed a PFS1/PFS0 ratio >1.0 in favor of experimental targeted therapy. As shown in Figure 2, targeted treatment was chosen according to the individual tumor profile and consisted of tyrosine kinase inhibitors, checkpoint inhibitors, growth factor receptor antibodies +/– endocrine treatment.
Response rate
Seven (33%) patients receiving targeted treatment experienced an overall response according to RECIST (Table 2). The disease control rate (DCR) was 62% (n=13). Out of 21 patients suffering from GI tumors, 7 (33%) patients had a partial remission, while 6 (29%) patients had a stable disease according to RECIST 1.1 criteria. Seven (33%) patients did not benefit from therapy and were progressive (one patient was still under experimental therapy and was not evaluated for treatment response at the day of censoring).

Full table
Survival data
The median PFS1 was 124 d (quartiles 70/193 d), whereas median PFS0 was 62 d (quartiles 55/83 d; P=0.014) (Table 3, Figure 3). The bootstrap 95% CI of the median of the PFS1/PFS0 ratio is 4−128. Median OS was 193 d (quartiles 115/374 d). The linear model showed that neither age nor gender had a significant influence on PFS1−PFS0.

Full table

Discussion
In this study we present a subgroup of the prospective clinical phase II EXACT trial to determine the efficacy of a MP-based therapy in pretreated GI cancer patients. Tissues derived from real-time biopsies of patients, refractory to standard therapy treatment, were characterized for their MP. Treatment suggestions were derived from the MP by the MDT. Out of 55 patients, 21 suffered from GI tract cancer. The median PFS of GI cancer patients under experimental treatment was 124 d and was significant longer than the median PFS upon the standard treatment immediately given before (62 d). Notably, 71% of patients (n=15) achieved a longer PFS in the experimental arm when compared with the immediately preceding treatment resulting in a median PFS ratio of PFS1/PFS0=1.59 (quartiles 0.43/12.46). When compared with other patients included in this trial, GI malignancies seem to be sensitive for molecular-based treatment concepts. Furthermore, shrinkage of the tumor upon the experimental treatment was observed in 33% of patients while the DCR was 62%. Tumor shrinkage in this late line setting is not common as the Recourse (15) and Correct (16) trials in metastatic colorectal carcinoma (mCRC) patients revealed. Finally, the median OS was 193 d (quartiles 115/374 d) which is a promising number for an experimental treatment after failure of standard treatment options.
In one of the first individualized treatment studies in cancer, von Hoff et al. reported beneficial effects of molecular profiling of patient’s tumors (17). Their study showed that 27% of the patients had a benefit from an individual targeted approach resulting in a significantly longer PFS when compared with the PFS of the previous regimen of the same patient (PFS ratio ≥1.3; 95% CI, 17%−38%; one-sided, one-sample P=0.007). Whether the genomic-guided approach to treatment results into beneficial outcomes for patients with GI cancer is still being studied and only few reports exist. By molecular profiling of 68 patients suffering from CRC with following targeted therapy in a clinical phase I trial, no clinical benefit was found with treatment of matched targeted agents (16). However, only few potential aberrations (KRAS, BRAF, PIK3CA mutations, PTEN and pMET expression) were considered in a retrospective setting from baseline biopsies. This also might not reflect the instable cancer genome especially when treated with targeted agents such as anti EGFR antibodies. The beneficial outcome in our study might be explained by the fact of progress in diagnostic techniques and particularly of the multitude of therapeutic options including immune checkpoint inhibitors.
Now, multiple promising targets have been applied with mixed results. Recently, vemurafenib failed to show beneficial activity as a single agent in BRAF V600E mutated patients (17). Actually, simultaneous blockade of the Wnt signaling pathway in combination with EGFR and BRAF inhibitors is investigated in an ongoing clinical trial (No. NCT022781333). Furthermore microsatellite instability (MSI)-high enhances immunogenicity due to neoantigen-specific tumor infiltrating lymphocytes. A phase II study already demonstrated clinical response with pembrolizumab by blockade of the PD-1/PD-L1 interaction in MSI-high patients (18). Patients with PD-L1 expression were also included in this study and showed a beneficial effect on PFS when compared with the treatment which was last given before. In gastroesophageal cancer, HER2 amplification was found to bear beneficial effects on survival. Patients who were treated with trastuzumab and combination-chemotherapy showed improved OS (median 13.8 vs. 11.0 months, P=0.0046) when compared to patients treated with chemotherapy alone (19). In addition, the two esophageal patients who showed beneficial effects in this study had a HER2 mutation. In consideration with the mixed results in different basket trials it becomes more and more important to search for those registered targeted anticancer drugs which showed beneficial results in previous studies. In this context, American Society of Clinical Oncology (ASCO) is developing the Targeted Agent and Profiling Utilization Registry (TAPUR) study which aims to facilitate patient access to marketed drugs which are predicted to be beneficial based on the analysis of patients’ genomic profile of tumors.
In contrast to the previous studies, the EXACT trial was not limited to a certain mutation but considered NGS, cytogenetic analysis as well as IHC profiling. However, this study has some limitations. The original EXACT trial was not designed to investigate GI malignancies but was open to all solid tumors. Furthermore, this prospective clinical trial was not randomized to a control group but considered patients as its own control as it was described previously (17). Finally, the sample size of GI cancer patients was rather small (n=21). However, we demonstrated that MP-based treatment decisions are feasible for extensively pre-treated GI cancer patients with certain characteristics to improve their prognosis upon failure of standard treatment options.
Although the number of patients is limited, the promising results of this study in combination with the progress in molecular technologies and increasing numbers of new targeted treatment options urge personalized medicine (PM) in the focus of future treatment concepts in cancer.
Conclusions
In GI malignancies the concept of PM was not investigated so far. In this work we describe PM as a promising approach for GI cancer patients who have no further treatment options after failure of standard therapy. Further prospective clinical trials with a higher number of patients are urged.
Acknowledgements
The study was supported by the Medical University of Vienna.
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Katsnelson A. Momentum grows to make ‘personalized’ medicine more " precise”. Nat Med 2013;19:249. [PubMed] DOI:10.1038/nm0313-249
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006. [PubMed] DOI:10.1001/jama.2014.3741
- Au TH, Wang K, Stenehjem D, et al. Personalized and precision medicine: integrating genomics into treatment descicions in gastrointentinal malignancies. J Gastrointest Oncol 2017;8:387–404. [PubMed] DOI:10.21037/jgo.2017.01.04
- Wong DJ, Ribas A. Targeted therapy for melanoma. Cancer Treat Res 2016;167:251–62. [PubMed] DOI:10.1007/978-3-319-22539-5_10
- Meric-Bernstam F, Brusco L, Shaw K, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol 2015;33:2753–62. [PubMed] DOI:10.1200/JCO.2014.60.4165
- Stockley TL, Oza AM, Berman HK, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med 2016;8:109. [PubMed] DOI:10.1186/s13073-016-0364-2
- Sohal DP, Rini BI, Khorana AA, et al. Prospective clinical study of precision oncology in solid tumors. J Natl Cancer Inst 2015;108:pii:djv332. DOI:10.1093/jnci/djv332
- Le Tourneau C, Delord JP, Gonçalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol 2015;16:1324–34. [PubMed] DOI:10.1016/S1470-2045(15)00188-6
- Stockley TL, Oza AM, Berman HK, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med 2016;8:109. [PubMed] DOI:10.1186/s13073-016-0364-2
- Schwaederle M, Zhao M, Lee JJ, et al. Impact of precision medicine in diverse cancers: A meta-analysis of phase II clinical trials. J Clin Oncol 2015;33:3817–25. [PubMed] DOI:10.1200/JCO.2015.61.5997
- Gomez-Martin C, Plaza JC, Pazo-Cid R, et al. Level of HER2 gene amplification predicts response and overall survival in HER2-positive advanced gastric cancer treated with trastuzumab. J Clin Oncol 2013;31:4445–52. [PubMed] DOI:10.1200/JCO.2013.48.9070
- Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008;26:374–9. [PubMed] DOI:10.1200/JCO.2007.12.5906
- Loupakis F, Ruzzo A, Cremolini C, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 2009;101:715–21. [PubMed] DOI:10.1038/sj.bjc.6605177
- Kieler M, Müllauer L, Koperek O, et al. Analysis of 10 adrenocortical carcinoma patients in the cohort of the precision medicine platform MONDTI. Oncology 2018;94:306–10. [PubMed] DOI:10.1159/000486678
- Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:303–12. [PubMed] DOI:10.1016/S0140-6736(12)61900-X
- Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909–19. DOI:10.1056/NEJMoa1414325
- Von Hoff DD, Stephenson JJ Jr, Rosen P, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010;28:4877–83. [PubMed] DOI:10.1200/JCO.2009.26.5983
- Dienstmann R, Serpico D, Rodon J, et al. Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials. Mol Cancer Ther 2012;11:2062–71. [PubMed] DOI:10.1158/1535-7163.MCT-12-0290
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74. [PubMed] DOI:10.1200/JCO.2009.23.3452